Novel study designs for rare diseases

Developing treatments for rare diseases poses many unique challenges that cannot be addressed by traditional study designs. Small populations will result in prolonged enrolment periods and/or inability to enrol sufficient number of patients to formally power clinical trials. Limited experience with the disease may result in lack of clarity regarding endpoints and key design assumptions due to limited pertinent clinical trials experience. There may also be ethical issues with the inclusion of a control arm (Beckman et al, 2022).
In 2023 the FDA published guidance on rare diseases. The guidance recognises that many rare diseases are serious conditions with no approved treatments. There is insufficient understanding of natural history of rare diseases due to the limited experience. They are highly diverse, and one development programme cannot be designed exactly like another. Rare diseases are therefore particularly affected by the challenges of traditional designs. Not only are sponsors less inclined to invest in the development of a product that will benefit very few patients, but enrolling a sufficient number of patients to satisfy requirements of traditional randomised clinical trials is often not possible.
The FDA guidance on rare diseases is making recommendations regarding several novel designs and approaches that can be used to address the development of treatments for rare diseases. Their examples include Bayesian methods, n-of-1 clinical investigations, randomised delayed-start designs, crossover designs, and master protocols. In this article, we will describe master protocols in some detail, and will then address some useful designs and approaches not mentioned in the guidance.
Master protocols
The FDA released a guidance in 2023, which defines a master protocol as a protocol designed with multiple sub-studies, which may have different objectives and involves coordinated efforts to evaluate one or more investigational drugs in one or more disease subtypes within the overall trial structure. Master protocols include umbrella trials, basket trials, and platform trials (Woodcock and LaVange, 2017).
Umbrella trials study multiple therapies that treat the same disease family in a single trial. Because they include multiple drugs, most umbrella trials involve multiple sponsors, and gaining agreement among them requires an initial investment in money and time.
Basket trials evaluate a single therapy for multiple diseases or disease subtypes that share a common molecular marker or other pathogenic feature, and hinges on a hypothesis that there is a strong link between disease, molecular target, and targeted therapy. In fact, basket trials may assume that the molecular pathway classification of disease is more fundamental than classification based on histology.
Platform trials are an investigational infrastructure to study multiple treatments, and/or multiple diseases within the same protocol, with a “platform” prepared in advance. This platform is an operational set-up that runs perpetually, such that compounds/diseases unknown today may enter the platform in the future. Compound or disease arms can also exit when there is sufficient level of evidence that they are effective or ineffective (Antonijevic et al, 2021).
There are many benefits of using master protocols. They lead to standardisation at the higher level, and saving in time and cost since operational infrastructure does not have to be recreated for each individual trial as in the traditional setting. In umbrella and platform trials there is the opportunity to share control arms which reduces the number of research subjects receiving only control therapy and reduces the overall sample size. Finally, master protocols naturally involve decision making in a broader context, which can result in great gains in efficiency.
Informational design
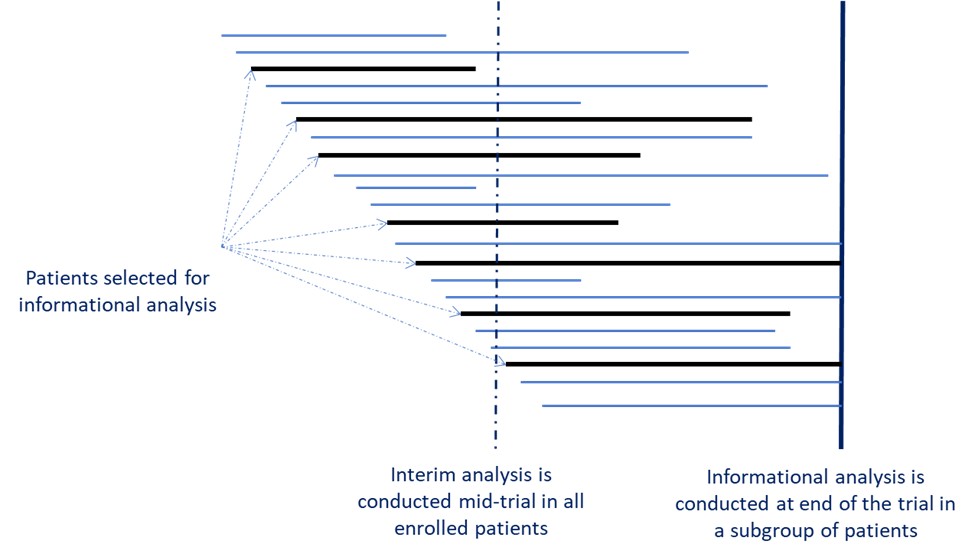
For many rare diseases, well-defined efficacy endpoints are not available. In this situation, the adaptive informational design (Beckman and Chen, 2019) can be utilised. In this design, a set of several candidate primary endpoints are pre-defined at the trial start, including the primary endpoint selection algorithm. A subset of patients – the informational cohort – is randomly selected prior to unblinding. Then, an analysis is run on this subset to select the best primary endpoint for the trial according to pre-specified selection criteria. Proper control of Type I Error must be provided.
Figure 1 illustrates the informational design, which is depicted by a solid vertical line. In this figure, patients randomised to be the informational cohort are represented with the black horizontal line. The dotted vertical line illustrates where the timing of interim analysis would be compared to informational analysis. Informational design would clearly be based on a lot more accumulated information than design with traditional interim analysis.
Use of external controls
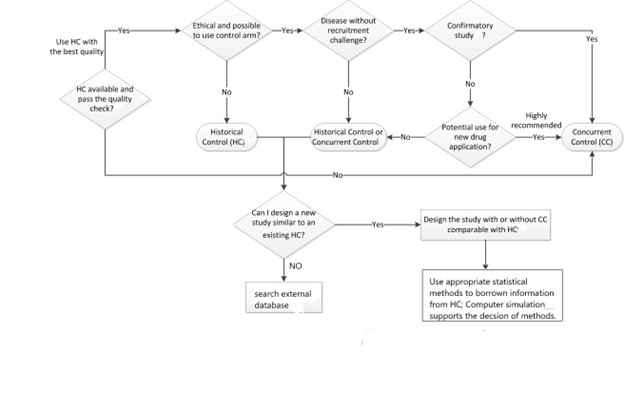
In developing treatments for rare diseases, it is important to consider utilising external control data to fully or partially replace a concurrent control, due to challenges to enrol sufficient number of patients in the trial. Additionally, external controls can address ethical concerns in recruiting patients with life threatening diseases with no credible control arm.
Figure 2 provides an algorithm for deciding when the use of external control is appropriate. (Tang et al, 2021).
In 2023, the FDA published the guidance for the use of external controls. One of the most important issues is the comparability of data. The following are factors that must be considered for the comparability:
- inclusion/exclusion criteria
- time period when external data was collected
- known prognostic factors
- diagnosis and prognosis
- treatment – formulation, dose, and frequency
- geographic region
- definition of endpoints
- handling of missing data and intercurrent events
In their communication with the FDA, sponsors must demonstrate why proposed data sources for the external control arm are fit for use as the external control. They also must provide reasons why the proposed study design is appropriate, including the operating characteristics. They will have to submit the statistical analysis plan (SAP) prior to initiation of enrolment. The SAP will have to demonstrate consistency in estimands, and methods for imputing missing data and handling multiplicity.
Developing treatments for rare diseases poses many unique challenges that cannot be addressed by traditional study designs. High level of design sophistication and flexibility is necessary to develop treatments for rare diseases in a timely and cost-effective fashion.
References
Antonijevic Z, Liu Y, Tang R, Huml J, Beckman RA, Mayer C, McMillan G. Patient Benefits from Innovative Designs in Rare Diseases. Rare Disease Drug Development: Clinical, Scientific, Patient, and Caregiver Perspectives. Springer; 2021 pp 147-160
Beckman RA, Antonijevic Z, Ghadessi M, Xu H, Chen C, Liu Y, Tang R. Innovations in Clinical Development in Pediatric Rare Diseases: Small Patients, Small Populations. Pediatric Drugs 2022
U.S. Food and Drug Administration. Master Protocols for Drug and Biological Product Development. December 2023.
U.S. Food and Drug Administration. Rare Diseases: Considerations for the Development of Drugs and Biological Products. December 2023.
U.S. Food and Drug Administration. Considerations for the Design and Conduct of Externally Controlled Trials for Drug and Biological Products. February 2023.
Tang R, Ghadessi M, Liu Y, Xu H, Chen C, Antonijevic Z, Beckman RA. Novel Approaches to Clinical Trials in Rare Diseases. Rare Disease Drug Development: Clinical, Scientific, Patient, and Caregiver Perspectives. Springer; 2021 pp 127-145.
Woodcock J, LaVange LM. Master Protocols to Study Multiple Therapies, Multiple Diseases, or Both. N Engl J Med 2017; 377:62-70. Doi: 10.1056/NEJMra1510062
About the author
 Zoran Antonijevic is vice president of statistical consulting at Bioforum. He held executive positions in pharmaceutical companies and CROs and designed well over 100 clinical trials in numerous therapeutic areas, many of which included innovative designs. Antonijevic was a long-time Chair and leader of the DIA Innovative Design Scientific Working Group. He has authored numerous papers, scientific presentations, courses and trainings, and was editor of the books “Optimization of Pharmaceutical R&D Programs and Portfolios” and, together with Bob Beckman, “Platform Trials in Drug Development”.
Zoran Antonijevic is vice president of statistical consulting at Bioforum. He held executive positions in pharmaceutical companies and CROs and designed well over 100 clinical trials in numerous therapeutic areas, many of which included innovative designs. Antonijevic was a long-time Chair and leader of the DIA Innovative Design Scientific Working Group. He has authored numerous papers, scientific presentations, courses and trainings, and was editor of the books “Optimization of Pharmaceutical R&D Programs and Portfolios” and, together with Bob Beckman, “Platform Trials in Drug Development”.











