When measuring European regulatory approval, the timestamp matters
Daniel Sanchez and Yin Ho explore the concept of cycle time in regulatory decisions in the pharma industry and their significance in market access. They consider the FDA compared with the EMA in this context and question whether we are comparing them fairly.
Cycle times for regulatory decisions are of great importance, especially in understanding market access for a drug. A cycle time measures the time from the date of submission, through date of approval. In the U.S., this is the time that it takes to get a drug approved by the FDA. The date of approval is generally the point in time when a drug is considered eligible for market access. In looking across the past four years of regulatory decisions, we noticed that some of the analyses involving the European Medicines Agency (EMA)'s cycle time used an inappropriate endpoint, which resulted in cycle times that looked two months shorter than they actually are. Adding an extra two months to market could make a tremendous difference, especially for life-saving drugs.
There has been a lot of discussion and interest lately about the length and speed of cycle times for drug development, specifically in comparing approval regulatory cycles for the Food and Drug Administration (FDA) and the European Medicines Agency (EMA). Last year we wrote a three-part series, "Life in the Fast Track" that looked into how the speed of previous FDA fast lanes compared with the EMA. One of the most interesting things we found was about how cycle times are calculated, and that they are not always measured the same way.
Understanding cycle times: are we all using the same math?
Cycle times calculate the time of approval for a drug. However, the time of approval of a drug may not necessarily be equivalent to the time of market authorization—which is a more important time point because it is when a drug becomes available to be prescribed.
When comparing the FDA and EMA, we found that many of the calculations that determine this relative speed have used time of approval and time of market authorization, interchangeably. This perspective is one that is driven from a lack of understanding about the difference between the two processes and represents an FDA-centric point of view.
Looking across the past four years of regulatory decisions by both the FDA and EMA, we found that some cycle time analyses published for the EMA actually used a different endpoint than the one that determines market authorization. Based on this discrepancy, we believe that some of the comparisons that determine the agency's cycle time or speed to market may be incorrect.
In the U.S., FDA cycle times are calculated from the time a drug is submitted for approval to when it is granted approval. Almost immediately after the FDA grants approval, the drug is authorized for marketing, and made available to the U.S. population. So for the FDA, the time of approval as the endpoint for market authorization is appropriate.
In Europe, the EMA's process involves one more step, creating an intermediate time point that seems to be mistakenly calculated as the point of market authorization for a drug in Europe. At the end of their evaluation process, the EMA puts forth an opinion for market authorization that is known as a non-binding Committee for Medicinal Products for Human Use (CHMP) recommendation—a non-binding opinion. This recommendation is subsequently passed to European Commission (EC), which according to the EMA "has the ultimate authority for granting marketing authorisations in the EU" and whose decisions tend to occur 2 months after the CHMP recommendation.1
What's interesting here is that in much of the analysis referencing EMA cycle times, it is the CHMP recommendation point that is used as the timestamp for market authorization. And while the CHMP recommendation "technically" marks the end of the EMA's evaluation cycle, the effective cycle time endpoint should be the time when the drug is granted market authorization to the European country populations—a point that is actually two months later! In essence, the endpoint that is used to calculate the cycle times for market authorization is an intermediate point in the process.
The real question here is whether we are comparing apples to apples when we say that FDA is faster than EMA. It also means that most regulatory watchers may not be looking at the appropriate endpoint for calculating and comparing relative cycle times.
Splitting hairs, or does this difference matter?
In most cases, the EC agrees with the CHMP's recommendation, but not always. From 2008–2011, there were at least three instances where the EC issued a marketing authorization in light of a negative CHMP recommendation, and another time where the EC refused to approve something despite a positive recommendation from the CHMP. Also, there were 11 drugs that were withdrawn from consideration by the applicant prior to an EC decision; despite a positive CHMP opinion. This may indicate the applicant's belief that the drug would not ultimately be approved by the EC. So while CHMP opinions and EC decisions are typically in line with one another, agreement is not a guarantee.
When looking at the relative speed between agencies, the use of this inappropriate endpoint of a cycle may create a not-so-valid comparison between EMA and FDA. The EMA's drug approval process is a two-step process, consisting of both the CHMP and EC's decisions. In comparison, the FDA is the only body that conducts regulatory review in the United States; therefore approval time is obtained by calculating the difference between the submission and marketing authorization dates. What this means is that using the CHMP date as an endpoint results in an artificial deflation of EMA cycle times by about two months—potentially a very significant difference.
We recently looked at the lag time between FDA approval and approval by other regulatory agencies for HIV drugs. Note the significant increase of lag time (dark blue bar) when the proper endpoint is used. Note: Time=0 represents FDA approval.
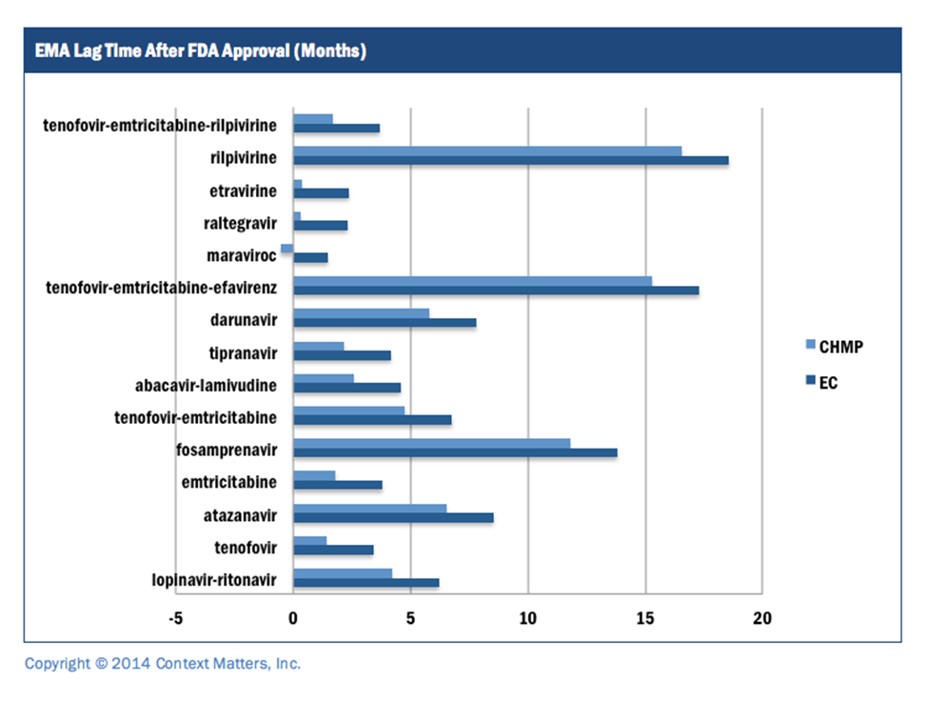
In this example, the FDA consistently (15 out of 15 times) approved a drug before its European counterpart. However, when using the appropriate EC endpoint (rather than the CHMP) to mark the end of the cycle time, the difference between FDA and EMA market authorization is actually two months longer.
While this is not a competition between agencies, there are real consequences to not recognizing a longer time between start of an evaluation and the availability of a drug to patients. Any analysis that uses this flawed endpoint can claim drugs are reaching EU member states two months faster than they truly are. The EMA already has longer cycle times than the FDA, and adding an extra two months to market could make a tremendous difference, especially for life-saving drugs.
That said, the EMA is more accepting than the FDA in terms of the types of data they receive for submission. Having more data to analyze could be one explanation for the relatively longer cycle times.
References

About the authors:
About Daniel Sanchez
Daniel Sanchez is in his second year at Context Matters as a Research Analyst. He received a Masters degree in Bioethics from New York University with a specialization in Health Economics and was awarded with Departmental Fellowship scholarship. Prior to that, he studied History and American Studies at Cornell University. He has worked at the Health Management Academy and the Montefiore Einstein Center for Bioethical Consultation. He is interested in the nexus of technology, healthcare, and regulatory processes.
About Yin Ho
Yin is the Founder and CEO of Context Matters Inc., a healthcare information and data analytics technology platform company based in NYC. A licensed physician trained in emergency medicine, Dr. Ho left the practice of medicine in 1998 to focus on the emerging eHealth sector. Prior to founding Context Matters, she was vice president of corporate strategy at Medidata Solutions (MDSO) – a global clinical trial software company – and was part of the executive team that took the company public in mid-2009. She has also held senior positions at Pfizer (PFE), leading initiatives and investments in eHealth, including electronic health records, electronic prescribing hubs, evidence-based medicine/comparative effectiveness policies, physician-payer relationships, and medical public policy. Yin has also served as director of business development at Emergingmed.com, an e-clinical trial company focused on cancer patient recruitment and advocacy, and a member of the advisory board of RapidTrials, Inc, which is focused on accelerating clinical trials through optimizing clinical investigator performance.
Yin is a dedicated New Yorker who is personally committed to making NYC a recognized capital of healthcare innovation. She co-founded NYC Health Business Leaders and is affiliated with numerous associations, including serving on the Advisory Board of NYC Economic Development Corporation's Bio and Health Tech NYC Entrepreneurship Lab Program, the Executive Committee for the Association of Yale Alumni in Medicine, and the Board of Directors of PS3 Afterschool (PS3til6) Program.
Yin received a BA from Brown University, MD from Yale University School of Medicine, and MBA from Harvard Business School.
Have your say: Are we comparing apples to apples when we say that FDA is faster than EMA?









