Gathering real-world evidence in the early access setting
Early access programmes offer the potential for gaining real-world evidence in the period between phase III and marketing authorisation, which could provide data to support the product's effectiveness and value proposition.
Evidence generation outside of the clinical trial setting remains a hot topic for pharma companies and regulators alike. With the advent of the European Medicines Agency's (EMA) adaptive pathways pilot, the UK's Early Access to Medicines Scheme and managed entry agreements, there is a particular emphasis on how evidence to support marketing authorisation can be gathered from outside the traditional routes.
Data mining from health insurance databases and from health provider databases is allowing researchers and companies opportunities to tap into large sources of patient-level data. However, many such sources are restricted by the fact that the data contained within them can be difficult to access, are usually country-specific, may be fragmented/incomplete and may not be capable of providing data about unlicensed medicines use.
Early access programmes (EAPs) (variously known as expanded access programmes, named patient supply, compassionate use etc.) represent a unique opportunity to gather real-world evidence – the first opportunity outside of the clinical trial setting – from a cohort of patients all receiving the same treatment, across multiple countries.
"A cornerstone of EAPs is that providing access to an unlicensed medicine must be driven by treatment needs only, not research needs"
A cornerstone of EAPs is that providing access to an unlicensed medicine through such mechanisms must be driven by treatment needs only, not research needs. However, within those confines, and given that any programme will have been put in place purely to satisfy unsolicited demands for treatment, it is perfectly legitimate to ask the participating physicians to provide data, so long as a) they are not asked to provide any data beyond that which they would capture as part of routine treatment of the patient, b) they are not obligated to provide data and c) they have the explicit consent of the patient to share that data.
Recent years have seen an increasing trend towards data capture during EAPs. Some of this data capture has been compliant, such as investigator-led efforts to collect data. However, the obvious downside of investigator-led data capture is the lack of control on the usage of such data by the pharma company, and lack of access to the actual data.
Early access real-world evidence within the continuum of clinical development
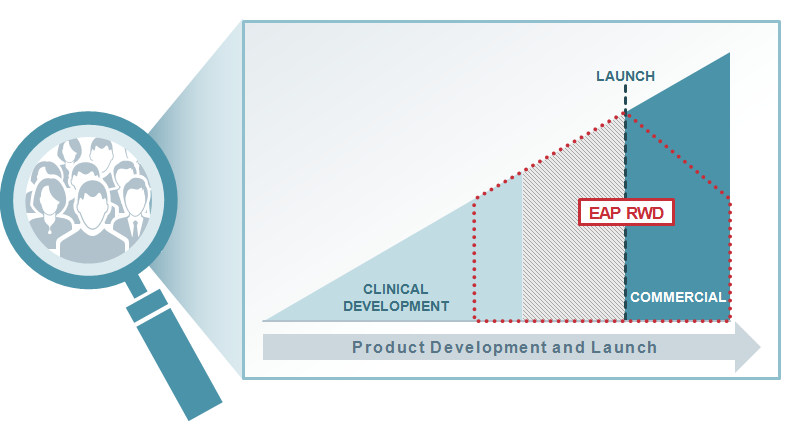
Another common approach is retrospective analyses of data from an EAP, with the obvious downsides that data becomes available long after the EAP, and generally after commercialisation, and fails to capitalise on the unique opportunities EAPs present – the ability to capture patient-level data during the crucial time between the completion of phase III and marketing authorisation.
A more worrying trend is towards 'hybrid' EAPs/open-label extension studies. Such an approach does not really fit with the 'spirit' of EAPs, in that there is a blurring of the line between providing access with a formal intention to conduct research. These two objectives should remain separate and, in cases where the need for such extensive data collection would warrant an open-label extension approach, an open-label extension should be conducted, but not under the auspices of an EAP.
However, there is now the potential to capture data prospectively during an EAP, without confusing research and treatment and in a less administratively burdensome way than conducting an open-label extension study. This new approach allows pharma companies to capture data they may wish they had captured during phase III, to examine signals which may have been identified during phase III, or to try to gain insight into efficacy outside the confines of a clinical trial, all during an EAP. Aside from insight into efficacy, such data, collected in advance of commercial launch, could help inform future studies and give insight into patterns of use and outcomes in the actual clinical setting, more reflective of the commercial-use setting.
"Data captured during EAPs should be viewed as supportive, ancillary data, which can provide insight and guidance"
There are some specific nuances around capturing data during an EAP. Typically, EAPs have a smaller, more heterogeneous patient population than a clinical trial population, but the intention of capturing data during an EAP should not be to replicate or obviate the need for further clinical studies, such as phase IV or open-label extension studies. The data captured during EAPs should be viewed as supportive, ancillary data, which can provide insight and guidance. For example, being able to track outcome measures in real-time in what may be thousands of patients, prior to commercial launch, has significant potential for pharmaceutical companies, even given the lack of robustness of randomised clinical trials.
Real-world evidence generation during an EAP can be a very strong tool when used appropriately. For example, in the rare disease setting, where patient numbers in clinical trials may be relatively low, an EAP presents an ideal opportunity to gather crucial data. Where there may be a lack of clarity over the therapeutic benefit of an Investigational Medicinal Product over a commercially-available product, data from an EAP could potentially help demonstrate effectiveness in the actual patient population reflective of the clinical setting. Such data could assist in demonstrating product value, potentially be utilised in Health Technology Assessments, supplement market access submissions, and may even have a role in adaptive licensing or managed entry agreements.
Compliant, prospective data capture during an EAP is an enormous, as yet untapped, resource, which has the potential to deliver significant benefits for both pharma companies and regulators alike.
About the author:
Dr Stuart Bell is Director of Strategic Services at Idis Managed Access, a part of Clinigen Group plc. With a PhD in vaccine development, he has a professional background in strategic consulting, market access, communication and marketing gained in pharmaceutical, biotech, governmental and non-governmental organisations across Europe. He leads a dedicated team of experienced consultants, helping Idis' clients gain the most benefit from their managed access programmes.
Read more from Idis:
Managed Access Programmes: maintaining treatment access post-trial









