FDA ‘breakthrough’ status – a new tool for building oncology brands?
Kelly Price looks at a new regulatory status and how Oncology Brands are using it to their advantage.
The FDA's new Breakthrough Therapy designation may offer oncology brands a powerful way forward – and some challenges for market research.
Phase III in oncology – between a rock and a hard place
The term 'Phase III' may strike terror in the hearts of oncology pharmaceutical executives – and rightly so. The clinical-trial process is supposed to separate the 'wheat from the chaff', with only those drugs most likely to succeed earning the right of further investment. Yet oncology products have a greater likelihood than other therapy areas to fail at Phase III (see graph) – well after the branding process has already begun and market expectations are somewhat set.
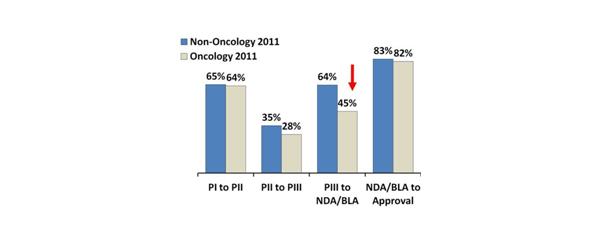
The spanner in the trial works is endpoints – in Phases I/II, the primary endpoint is usually PFS, which is easier to demonstrate in the short term. However, in Phase III the goalpost shifts to OS, and the playing field is suddenly not so level. Demonstrating OS is difficult enough in a disease that mutates as fast as you can throw new weapons at it. When you combine tricky trial design issues, the waters are muddied all the more.
Oncology is orphaned
One way that pharmaceutical companies have mitigated these risks is to pursue orphan indications for products' initial launches. The Orphan Drug Act of 1983 grants special status to drugs developed for diseases that affect 200,000 or fewer individuals. On initial inspection, it might seem strange that companies would target such limited markets, but Orphan Disease status confers significant benefits, including subsidies for new research, exclusivity, slightly shorter approval times, and waived regulatory fees.
Orphan drugs now constitute a market worth $50 billion, with a 5% higher growth rate than non-orphan therapies. Oncology products in particular have made use of this strategy, and currently over 40% of orphan drugs are indicated to treat various cancers. In fact, some of the top grossing oncology drugs – 'household' brand names like Rituxan, Herceptin, Avastin, Gleevec and Revlimid – have orphan status.
The recent shift towards 'personalized' medicine in oncology has reinforced this strategy by sub-segmenting tumor indications down to the molecular level. The result is drugs like Pfizer's Xalkori, which is indicated for metastatic non-small cell lung cancer patients who harbor the ALK mutation – in other words, approximately only 3–5% of patients with this particular type of lung cancer.
Once approval for an orphan indication has been secured, the strategy has been to branch out to other indications. This explains why Nexavar, Sutent, Afinitor, and Votrient all launched first in renal cell carcinoma, a tumor with an incidence of less than 60,000 in the United States, before pursuing indications in other tumor types.
Orphan status also confers excellent pricing strategy advantage. Given that Orphan Diseases are by definition niche indications affecting small populations, companies have been able to charge higher unit prices for these drugs, backed up by the argument that they need to re-coup their costs in a limited market. Payers in turn have been willing to accept higher prices, knowing that the patient pool that will need the drug is limited. As a result, we have seen drugs for increasingly chronic orphan indications such as Gleevec in CML charging up to almost $100,000 per year per patient.
However, this has proven to be a double-edged sword as the number of orphan drugs has swelled along with their prices. A group of over 100 top US oncologists recently called on companies to cut their prices, and payers are beginning to demand just that.
Breakthrough status – a new strategy
Just as the pressure to cut prices is building, a new strategy has emerged to help mitigate risk: breakthrough status. Last July, the FDA introduced a new process to speed up approvals for products that show initial promise of 'substantial improvement on existing therapies for clinically significant endpoints'. The beauty of this designation is that it can be secured on Phase I/II data, since the goal is to get these products to patients faster. Breakthrough status confers a number of benefits: FDA hand-holds companies through subsequent trial design, and products receive automatic 'fast track' designation. In addition, trial design is 'streamlined' so that fewer patients are exposed to the comparator arm.
"A group of over 100 top US oncologists recently called on companies to cut their prices, and payers are beginning to demand just that. "
The financial upside for companies are potentially huge: less money and time spent on large trials, significantly faster approval, and no restriction to small indications. As a result, breakthrough products can enjoy a longer patent life in a larger market for a potentially lower investment. In other words, it's a win-win-win.
A 'breakthrough' benefit for oncology branding
Oncology products have been the biggest beneficiary of breakthrough status so far - of the 10 approvals to date, the majority have been for cancer drugs. And in terms of oncology branding, it makes perfect sense. Breakthrough status is a far better branding strategy than orphan disease designation: the latter speaks to the therapy area, implying that it is limited and neglected and desperate for any therapy that might work; the former, by contrast, is inherently positively linked to the product itself, creating a halo that feeds directly into positioning. It says, "This product is a real advancement over existing therapies. It's one to watch!"
Investors seem to have taken note: in February, Pharmacyclics' ibrutinib was the first oncology product to receive the designation and the company's share price saw a 13% jump in days after the announcement. Similarly, Genmab's stock rose 10% on the announcement that its daratumumab had been approved as well. One company is already using breakthrough therapy status application itself as positive PR: Novartis boldly announced on May 6th of this year that it will take advantage of breakthrough status for its product LDK378 – days before said status was granted.
Faster and faster – implications for brand planning
Novartis has claimed that under breakthrough status, LDK378 may be commercially developed within 3 years – an unheard of speed for an oncology product. With far less time for brand planning and launch preparation, fewer studies can be conducted, and those that are conducted must be done very quickly, often having to cover multiple objectives at a time. One potential downside of these truncated timelines, however, is that more risk is introduced as decisions are taken in the absence of data. Market research agencies would do well to adapt to the new 'breakthrough' reality by developing thorough quick-turnaround research studies to aid pharmaceutical companies in their branding process. That would be a win-win-win-win.

About the author:
Kelly Price is Head of TPSi's Oncology Specialist Group at THE PLANNING SHOP international.
With a background in strategic consulting, she has over 10 years' experience in oncology-specific research across the vast spectrum of tumor types.
You can contact Kelly on +1 215 680 8720 or at kelly.price@planningshopintl.com
Is breakthrough status a far better branding strategy than orphan disease designation?









