Better outlook for clinical trials in India
When the Indian government changed the guidance around conducting clinical trials, it put the country out of step with the rest of the world and led to a dramatic drop in numbers of approvals. Gopal Pai explains what the problems were and what changes have been made since to improve the situation.
A year ago, the clinical research environment in India was set back drastically by government guidance changes, which caused concern for the global research community looking to conduct clinical research projects there. Their fears related to perceived additional risks around serious adverse events (SAEs) causing death and injury, plus related compensation/medical reimbursements which were not in line with wider international standards. Even the country's insurance industry could not support some of the amendments.
Following urgent discussions, these guidelines have been revisited and changed to align more with global standards and timelines, which has helped the sector recover in the last few months.
The damaging changes in July 2014 set the industry reeling as they contributed to the numbers of approvals plummeting from 500+ pre-2011 to 25+ in 2013-14.
The recent amendments will help the trials environment improve further, but numbers will still be low owing to specific conditions, based on local benefits vs risks, that will influence the decision of the regulators when they make a 'go' or 'no-go' decision.
The two important aspects that have now been rolled back to global standards are the safety reporting timelines and the compensation related to SAE death and injuries related to the study drug. These new guidelines were implemented in June 2015.
The first outcome after the debates on the July changes was an order issued in September 2014, stating that all the global clinical trials should be evaluated based on three parameters, to avoid unnecessary trials being conducted in India:
• Assessment of risk versus benefit to patients;
• Innovation vis-à-vis existing therapeutic option;
• Unmet medical need in the country.
This is now the most important summary that must be incorporated with the regulatory approval documents. It is pivotal to a study being evaluated by the Subject Expert Committee (SEC), within the Drug Controller General of India's (DCGI) approval process.
It will ensure a good level of transparency around the decision-making process on global clinical trials being approved by the regulatory agencies. If there are no strong arguments in favour of the benefits to the Indian population and the drug's impact on the above three elements in the trial, it should be assumed that the regulatory agency will not provide approvals unless a more convincing medical rationale is provided on the advantages to citizens and community health statistics.
Roll-back of SAE reporting timelines
Previously, the safety reporting timelines specified that the investigator must report any SAE within 10 days of its occurrence, as per the guidance, GSR 53(E), dated 30 January 2013. This requirement was a major challenge as it conflicted with all the international guidance and database tracking systems and regulatory timelines, which specified 14 days. The new version of the guidance has brought India into line and now states 14 days too, vide the new guidance, GSR 889(E), dated 12 December 2014.
Changes to safety reporting submission workflow and timelines
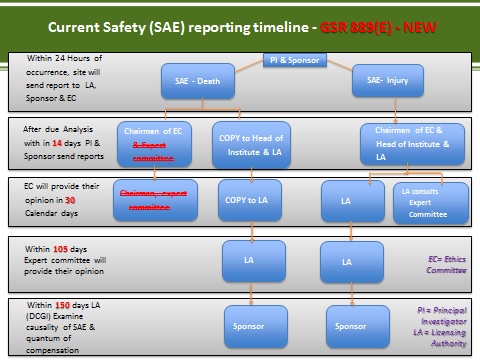
Alongside this, the following reporting steps were also changed so the investigator no longer sends the reports to the Expert Committee directly. This is now managed internally by the DCGI's Safety section and reported to the Expert Committee, as required. See the submission workflow in Figure 1.
Figure 1
Changes to SAE-related compensations and free medical management
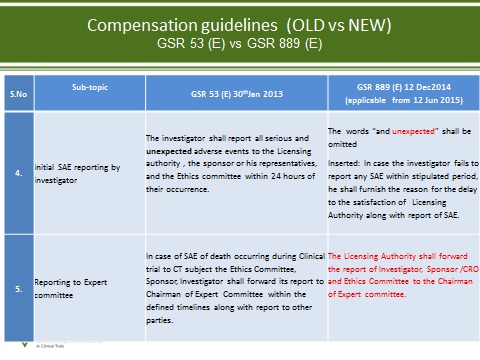
The changes to the other clauses clarify the circumstances around compensation and free medical management. A comparison of the changes is presented in Figures 2 and 3.
Figure 2
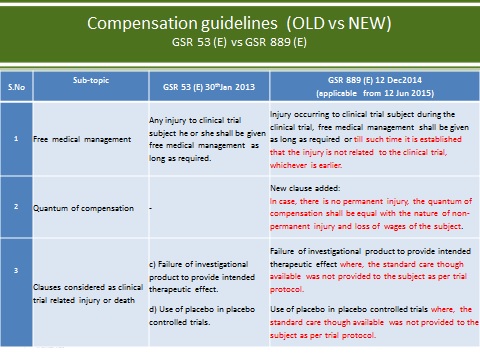
Figure 3
Pre-submission Meeting with regulatory agency
ON 28 January 2015, India's Central Drugs Standard Control Organisation (CDSCO) released a Notice on System of formal Pre-submission Meetings (PSM) of applicants with CDSCO (optional).
As per this notice, the regulatory pathways for applicants will be simplified, enhancing:
• Transparency
• Accountability
• Predictability
• Speedy disposal of cases.
Draft standards
On 3 March 2015, the CDSCO office released draft standards for accreditation of sites, investigators and ethics committees, along with the application format for accreditation.
These standards were prepared by the National Accreditation Board for Hospitals and Healthcare Providers (NABH) and the Quality Council of India, in consultation with various stakeholders. The draft set out the criteria for the accreditation of ethics committees, investigators and sites.
These standards are not mandatory yet, but it is anticipated that over the next few years, all the key parties will be made accountable and their operational standards improved as a result of these efforts by the country's regulators.
About the author:
Gopal Pai PhD is general manager at George Clinical India. He holds a postgraduate degree in Microbiology and a doctorate degree in Applied Biology from Mumbai University. He has formal business management training and over 25 years' experience in pharmaceutical research and development.
Read more on clinical trials:









