Reinventing drug invention
So…where are all the new drugs?
Superbugs haunt hospitals worldwide, killing millions and costing billions in healthcare spending. To this day, infectious diseases continue to literally plague the developing world, a slow-burning humanitarian disaster. COVID taught the developed world that infectious diseases do not respect national borders, but the US and Europe also face direct threats: a homegrown problem of antibiotic resistance that has been festering for years.
Ignoring these growing risks is short-sighted. So far, life-threatening infections remain relatively rare compared to the pre-modern era. However, it’s possible that within our lifetimes the healthcare system we take for granted could turn back into the medieval gauntlets of infection and mortality that they were prior to modern antibiotics. The impact on healthcare spending is already enormous: by one estimate, C. difficile infection alone already costs US hospitals more than $8 billion annually, which was an incredible 2.3% of all US hospital spending in the year studied (2009). That’s just for one bacterium; and the problem has only grown since then.
Why aren’t biopharma companies creating new cures for these diseases?
Diagnosing the problem
Sadly, the market for new anti-infectives is badly broken, and there are many causes. To begin with, no other field of medicine is more sensitive to rising development costs than infectious disease. The drugs for treating most common infections are already very good, so it’s difficult to match or beat that high level of efficacy as required for FDA approval. And because the most prevalent infections were tackled first, the remaining ones tend to represent ever smaller addressable markets. With every new approval, the available market space for a new product shrinks further.
In the face of shrinking markets and rising costs and risks for new anti-infectives, it's no surprise that pharmaceutical companies and investors shy away from investing in such drugs. Seeing no way to recoup the cost and risk of investing in new medicines for these diseases means that drug development for these diseases is hostage to whatever government and charitable funding happens to be available - a small fraction of the private sector’s R&D budget.
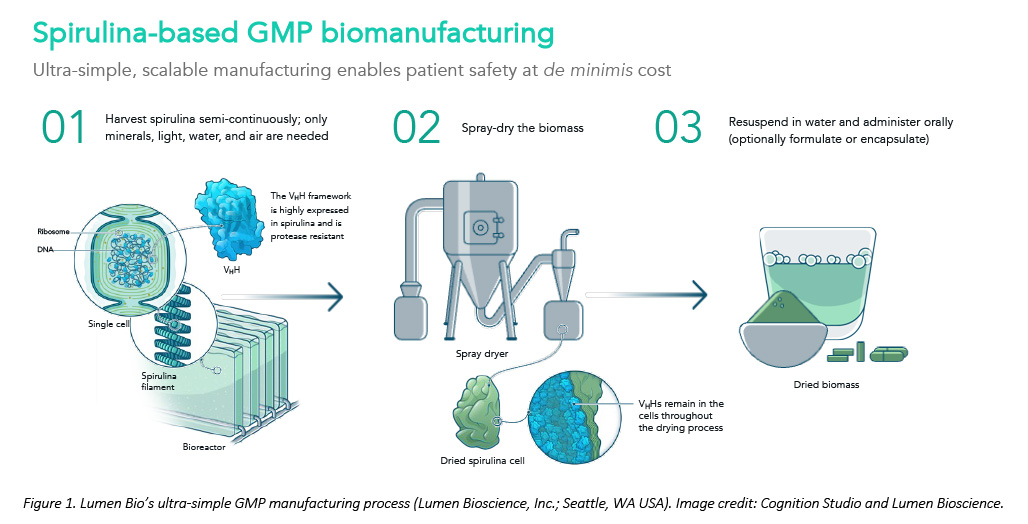
The forever war against human microbial pathogens desperately needs innovation, but fresh thinking is needed to restore the R&D pipeline. Thanks to a serendipitous meeting with the Gates Foundation, spirulina-based biotherapeutics are one way to slash development costs and timelines for new anti-infectives (see Figure 1). The technical details on how - and why - this platform works were featured recently on the cover of the journal Nature Biotechnology. Below we share three key lessons learned that can help other drug developers bridge the world’s gaping shortfall in new anti-infective drugs.
Cutting the cost of late preclinical development
It is not widely appreciated that late preclinical development is often the most expensive stage of the drug development pipeline once clinical attrition and the time-value of money are properly accounted for, particularly for biologics. This surprises a lot of people in the industry, but the results have been independently replicated by various researchers.
The main issue is complexity. For a conventional injected biologic drug, each new drug candidate requires expensive specialised equipment (stainless steel fermentation tanks), growth media (including a range of pricey growth factors and nutrients), and labour (especially to establish and maintain sterility). The cell lines themselves are finnicky, so getting them to behave stably in a large-scale manufacturing process requires intensive optimisation work. Downstream processing - where the drug is purified and formulated for injection - is even more complex and costly.
A spirulina-based GMP process, however, can simplify things. Because spirulina GMP cell lines are more stable and easier to engineer, cell-line establishment is much faster and cheaper. The growth media is a simple salt-water mix, and sterility is not required because the biomanufacturing host organism (spirulina) is a photosynthetic extremophile with a preferred growth medium hostile to most potential contaminants. Spirulina is a safe mass-market food (i.e., “GRAS status”) so downstream purification is not required for oral therapies. Even for intranasal applications, relatively little handling is required.
The direct savings (both time and cost) are profound in their own right, but more importantly these efficiencies enable new kinds of therapeutic concepts that would otherwise be commercial non-starters.
Fighting variants with valence
Infections involve broad natural diversity. SARS-CoV-2 has many variants after just four years with humans. Older pathogens like influenza and infant diarrhoeal disease have even greater diversity from attacking us for decades or centuries.
Yet, almost all biologic drugs today are monotherapies: a single molecule to cover the entire disease. Part of the remarkable success of antibiotics in the 20th century is down to the fact that they worked across a wide range of bacterial infections, albeit often with significant side effects. Modern antibody drugs tend to be safer, but more targeted, unable to handle diverse pathogens. Covering all the variants has bedevilled would-be makers of antibody drugs for Covid-19.
The time and cost savings in late preclinical development of spirulina-based, mucosally delivered biotherapeutics makes it feasible to create multi-antibody “cocktails” that cover this natural diversity. For some infections, this is straightforward. For example, with C. difficile infection, a three-antibody cocktail can cover the entire known universe of TcdB toxinotypes. In a neat twist - and some clever math - an artfully designed antibody cocktail can improve drug potency at the same time, an effect called “synthetic avidity”.
For some diseases, this is harder. For example, a programme directed at developing world infant diarrhoeal diseases backed by the Gates Foundation, DoD, and CARB-X requires a cocktail of 12-20 monoclonal antibodies to cover all strains of enterotoxigenic E. coli and C. jejuni. Such “high-valence” antibody cocktails are theoretically possible, but wildly unaffordable with conventional biomanufacturing. Yet, a spirulina-based programme is now in the clinic.
Rethinking delivery to improve safety and efficacy
Most drugs available today are designed for delivery to the blood serum, typically through injection. This holds true for all antibody drugs on the market. This approach has advantages, but there are trade-offs. Most importantly, systemic delivery exposes every tissue in the body to the drug. This increases the risk of side effects and unexpected toxicity.
Worse, because antibodies are very large molecules, they diffuse out from the blood stream into interstitial tissues at a very low rate. Compared to serum concentrations, antibody drug concentrations in these tissues are reported to be just 1% or less.
This makes it difficult for enough antibody drug to get to the site of infection because most pathogens attack us through our mucosal tissues (GI tract, nasal passages, etc.) and other topical surfaces. Very few are like malaria, which is carried about and delivered to the blood serum by mosquitoes. Because efficacy is concentration-dependent for nearly all drugs, this partly explains why so few antibody drugs have been developed for infectious diseases. It also goes a long way towards explaining why the few successes come in abnormally high dose sizes.
Biologic drugs for mucosal and topical delivery not only improve product safety (because antibodies are too large for intact systemic absorption by the GI tract), but also promise to enhance product efficacy (concentrating the antibody drug in the mucosal tissues, which are most directly attacked by most common pathogens). Better yet, unexpected safety and tolerability issues are the leading cause of clinical trial failure, so greater expected safety helps drive down development costs by reducing clinical attrition rates. It’s a triple win.
Conclusions
Making a new drug is hard. Doubly so in the field of anti-infectives, where the existing drugs are already pretty good and the addressable markets are relatively small. These two factors alone largely explain why investors and big pharma rarely support new investment in this field today. Significantly reducing the development costs is key to making such programmes investable again. The basics are all worked out; what’s needed now are more ideas for how to deploy this new technology in the real world.
About the author

Brian Finrow is founder and CEO of Lumen Bioscience, a clinical-stage biotechnology company in Seattle.










