Best practice for monitoring medication errors
Seema Jaitly defines what constitutes a medication error and explains the process for accurate recording and reporting so that risks can be assessed and reduced.

Seema Jaitly
There is a need for specific monitoring of medication errors as these are risks that are usually preventable. Looking at the public health impact of medication errors, inpatient medication error rates of 4.8% to 5.3% have been reported 1 and it has been estimated that 19.7% - 56% of all adverse events (AE) occurring in hospitalised patients are due to medication errors that could be preventable.
The definition of a medication error takes into account the unintended failure in the drug treatment process that leads to, or has the potential to cause, harm to the patient. It does not include intentional acts, such as intentional override or off-label use 2.
Medication errors can be classified depending on when in the process they have occurred. This could be during prescribing, storing, dispensing, preparation or administration of the product. Examples include: the wrong product being prescribed to a patient because it sounds the same as another product for a different indication; the wrong product dispensed because its name is similar to that of another product, or the wrong product taken by the patient because they cannot recognise their tablets adequately or have misunderstood how they should be taken.
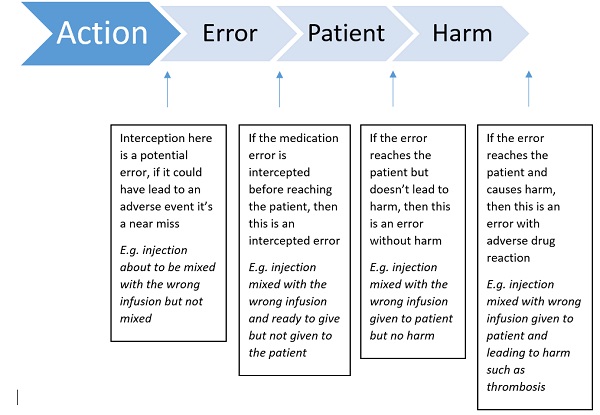
Figure 1. Flow diagram of types of errors (adapted from EMA guidance)
It is essential that there are mechanisms to recognise specific patterns of medication errors occurring, so rapid action can be taken to minimise these preventable risks. This is where management of information coming in is essential.
The European Medicines Agency's (EMA) 'Good practice guide on recording, coding, reporting and assessment of medication errors' came into effect on 27 November 2015. This guideline states the various sources of information relating to medication errors, such as spontaneous reports, literature and solicited reports (non-interventional studies). These can have adverse drug reactions (ADRs) associated with them or be a report of medication error with no AEs.
Those with ADRs need to be assessed for regulatory reporting, whereas medication errors without ADRs do not need to be reported in an expedited manner, but they do need to be recorded and available for analysis and summary in the Periodic Benefit Risk Evaluation Report (PBRER), Risk Management Plan (RMP) and for signal detection purposes.
Correct coding
It is therefore important that these cases are coded correctly so that they can be retrieved from the safety database. The Medical Dictionary for Regulatory Activities (MedDRA) should be used as the coding dictionary and the terms relevant for medication errors sit under the system organ class – Injury, poisoning and procedural complications. The primary stage of the error should be identified and coded, as should all the subsequent stages reaching the patient, e.g. prescription as a primary stage and, if it reached the patient, dispensing and administration should also be coded, so that all stages of the error can be retrieved. There is a preferred term (PT) of intercepted medication error and also one for circumstances or information capable of leading to a medication error that can be used to ensure correct codes are used.
Consistency
A standardised MedDRA query (SMQ) is being developed, which will provide a consistent way of retrieving medication error cases from the safety database. Additionally, it is important that medication errors are followed up to ensure that they can be classified appropriately and that the stages of the process can be recorded accurately. If there are significant issues related to confusion with names for centrally-authorised products, an email should be sent to the EMA's name review group at nrg@ema.europa.eu. Should any medication errors meet the definition of an emerging safety issue, an email should be sent to P-PV-emerging-safety-issue@ema.europe.eu.
Patterns
Accurate coding is essential as this is the one way that patterns of medication errors can be shown. For example, if there are 100 medication errors and 50 of them are related specifically to preparation and are related to a product being mixed up with incorrect infusion fluid then this would be an area to review to assess why these errors are occurring. For these patterns to be picked up the preparation would need to be coded so this can be seen; otherwise there would be a need to review every single case and subcategorise them.
Within the Periodic Safety Update Report (PSUR)/PBRER and risk minimisation plan you are required to provide a summary of medication errors. In order to do this properly the data relating to medication errors must be analysed. When reviewing the data for a PSUR it is useful to sub-categorise the individual cases with further details in a spreadsheet so errors can be grouped together and discussed in a logical manner. If there are cases of unintentional overdose with an oral solution, this could be due to the dropper being defective or the dropper being hard to read or the instructions not being clear. It may be hard to code for all these things specifically if the codes are not available. What you will retrieve from the database is: medication error, unintentional overdose, and there will be a need to review the case narrative to fully understand the root cause issue. This will then enable an assessment to be made about the need for any label or product changes to minimise this specific risk.
The EMA published another good practice guide on risk minimisation and prevention of medication errors at the same time as the first good practice guide. As most medication errors are preventable, risk minimisation is key. Generally the approaches are similar to other risks. However there are specific areas that need to be considered for this risk area. These include aspects such as the product name, to ensure no confusion, concentration and strength of product, and ensuring no confusion if there are similar products on the market with different strengths, as well as route of administration if there are specific instructions needed.
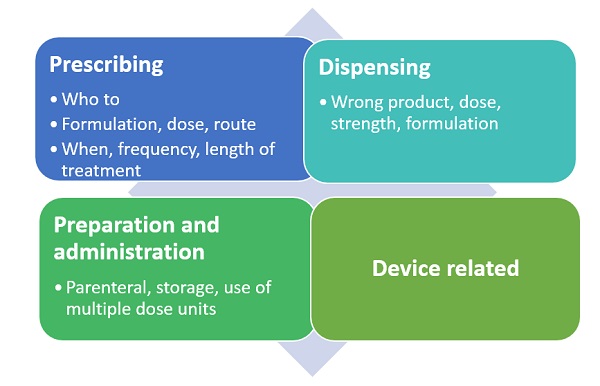
Figure 2: Possible medication errors in the different process areas
Risk minimisation consists of routine and additional risk minimisation. Routine refers to standard labelling, pack information and pack size limitations, whereas additional risk minimisation refers to any activities additional to these. Provision of educational materials is the most common additional risk minimisation activity for medication errors and these are usually used to ensure health care professionals are aware of specific instructions for prescribing, dispensing or administration.
High-risk groups
There are specific, high-risk groups that need to be considered for medication errors. For paediatric patients, points to consider include ensuring that it is clear from what age the product can be used, plus dosing calculations, which can be complicated if dosing is based on body surface area, for example, rather than a fixed dose.
For elderly patients, polypharmacy and the possibility of patients receiving contraindicated medication unintentionally should be considered, as well as issues with patients being able to read the information that comes to them.
Patients with visual impairment are most likely to need eye drops and the font size needs to be examined to avoid medication errors. Consideration should be given to the use of braille.
As with all risk-minimisation activities, the effectiveness of the activity needs to be assessed with well-defined process and outcome indicators to measure against for further risk minimisation. National reporting systems that collect data on medication errors and outcome data can be used for this, as can spontaneous reporting, which is the most common. Occasionally a post-authorisation safety study may be required especially following HCP communication or label changes to ensure these have had the desired effect, and sometimes a survey can be performed.
References
1 Wittich, Christopher M et al. Medication Errors: An Overview for Clinicians. Mayo Clinic Proceedings, Volume 89, Issue 8, 1116-1125.
2 GVP Module VI, Management and reporting of adverse reactions to medicinal products (Rev 1). 16 Sep 2014.
About the author:
Dr Seema Jaitly qualified in Medicine from Charing Cross and Westminster Medical School in 1992 and worked in hospital medicine for four years. She has worked in the pharma industry for over 18 years at CROs and companies spanning clinical research, medical affairs, pharmacovigilance and the EU QPPV role. In 2010 she founded Essjay Solutions to offer pharmacovigilance services, consultancy and contracting services.
She is currently studying for an MSc in epidemiology with the London School of Hygiene and Tropical Medicine.
Read more from Seema Jaitly:










