Regulatory timelines in the Asia-Pacific

An at-a-glance round-up of the arrangements for clinical trial applications in 10 countries in the region.
Australia

Australia is unique in that its Human Research Ethics Committees (HRECs) have responsibility for the oversight and ongoing monitoring of trials, including reviews and approvals. The first step is submission of the clinical trial to the HREC for approval, followed by regulatory notification to the Therapeutic Goods Administration (TGA). Additionally, public hospitals are required to undertake a research governance step in the form of a site-specific assessment (SSA) for each project. Therefore, for public sites, HREC and an SSA is required for a clinical trial to commence.
The most common approval process for clinical trials using unapproved medicines in Australia is via the Clinical Trial Notification (CTN) system. Australia also utilises the Clinical Trial Exemption (CTX) scheme. The CTX scheme involves the sponsors submitting an application to conduct one or more clinical trials with a particular investigational product to the TGA for evaluation and comment. However, as the CTN path is preferred, this scheme is rarely used.
The TGA does not review any data relating to the clinical trial. The institution or organisation where the trial will be conducted – the 'approving authority' – gives the final go-ahead for conducting the trial at the site, having taken into account advice given by the HREC. CTN research projects must not start at a site until the research project and investigational site have been notified to the TGA and the appropriate notification fee paid. It is preferable to wait for the TGA to confirm receipt of the CTN before recruitment commences.
India

Pharma companies wishing to conduct clinical trials in India are required to obtain written permission from the Drug Controller General of India (DCGI). The DCGI is part of the Central Drugs Standard Control Organisation (CDSCO) looking after the biopharma industry and responsible for approval of licences for specified categories of drugs, such as blood and blood products, intravenous fluids, vaccines and sera. CDSCO has developed an Online Clinical Trial Application and Monitoring System (OCTAMS) for submission of clinical trial applications to DCGI. Once OCTAMS is fully operational and CDSCO provides a directive, clinical trial applications can be submitted online as an alternative to physical submission of hard copies. The study site is responsible for the initial submission of the documents and responses required by the independent ethics committee (IEC). The IEC submission is made under the requirements of the local committee. The application can be made in parallel to a standard DCGI application, but India does not have a central ethics committee so each Investigational Product has to get approval from an institutional EC accredited by DCGI in the same city as the site. Further, EC submissions can occur in parallel with the regulatory submission of clinical trial application.
Singapore
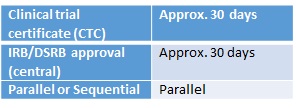
A Clinical Trial Certificate (CTC) must be applied for in order to undertake clinical trials on both unregistered and registered local pharmaceutical products. Applications for a CTC must be made online to the Health Services Authority by the sponsor (or delegate), which should be a locally-registered company. This application can be made in parallel with Institutional Review Board/Domain Specific Review Board (IRB/DSRB) applications. Regulatory approval is issued independent of the IRB/DSRB approval. The submission to the IRB/DSRB can be done in parallel to the regulatory submission, with the regulatory approval not contingent on ethics approval.
There are two IRB/DSRB approval processes: the IRB is used by private hospitals, while the DSRB is used by government hospitals for both single-site and multi-site applications. Regulatory approval is issued independent of the IRB/DSRB approval. Electronic submission is preferred.
Malaysia

Before commencing clinical trials in Malaysia, clinical trial applications can be submitted to the Drug Control Authority (DCA) in parallel with an Ethics Committee (EC) submission. However, approval from the National Medical Research Register (NMRR) is required to initiate the Clinical Trial Import Licence (CTIL) and the Medical Research and Ethics Committee (MREC) or Independent Ethics Committee (IEC) review. A CTIL is needed for an imported Investigational Product (IP) and a Clinical Trials Exemption (CTX) is needed for a local IP. Submission of the ethics approval is required prior to the final release of the CTIL or CTX approval. The application for a CTIL or CTX may be made in parallel with the IEC/Institutional Review Board (IRB) submission. However, the approval letter from the IEC (for a multi-centre study, approvals from all IEC/IRBs) must be submitted to the DCA as soon as it is available and the CTIL/CTX will not be issued prior to IEC/IRB approval.
Any research involving human subjects in Malaysia must get written approval from the EC. Malaysia has two EC bodies: Central EC for Ministry of Health (MOH) hospitals and institutions (MREC). If a hospital/institution does not have a local IRB/IEC, that site will be submitted to the Central EC. There are also local ECs at various teaching hospitals and private hospitals/institutions.
South Korea
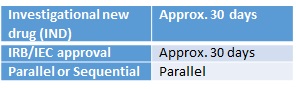
The MFDS (Ministry of Food and Drug Safety) offers a pre-investigational new drug (IND) submission consultation, which is advised prior to making the IND submission. There is no expiry date for MFDS approval, but trials are to be initiated within two years of approval. Additionally, Korea also follows the Korean Good Clinical Practice guidelines (KGCP), which is harmonised to the International Conference on Harmonisation Good Clinical Practice (ICH GCP) guideline. Generally, the IRB/IEC in Korea are local. However, a few hospital groups operate under a central IRB/IEC. The IRB/IEC process can take place in parallel with the IND application, for which all documentation should be in Korean. However, MFDS approval must be received before research commences.
Hong Kong
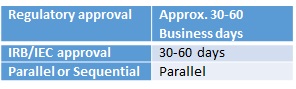
A Certificate for Clinical Trial/Medicinal Test is required for conducting clinical trials on human beings, or a medicinal test on animals, in Hong Kong. The Pharmacy and Poisons Committee, established under the Pharmacy and Poisons Board of Hong Kong, is the statutory body to issue the Certificate. It issues it through two mechanisms, namely, the Standard Scheme and the Listed Scheme. When the sponsor of a clinical trial is a pharmaceutical company or research organisation/institution, the application should be submitted under the Standard Scheme. An application for a medicinal test should also be submitted under the Standard Scheme. For clinical trials which are initiated and conducted by an investigator acting as the sponsor, the Certificate application may be submitted under the Standard or Listed Scheme. Application to the IRB/IEC can be made in parallel with the regulatory submission, but the final regulatory approval will not be granted until IEC/IRB approval has been received.
China
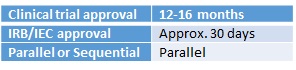
For clinical trials to be undertaken in China, a Clinical Trial Application must be made to the China Food and Drug Administration (CFDA) (imported Investigational Medicinal Product [IMP]) or provincial FDA (local IMP), in the approved CFDA format. For an imported IMP the National Institutes for Food and Drug Control will undertake a registration inspection and organise a technical review with the Centre for Drug Evaluation (CDE). This inspection will occur in parallel with the CFDA review of the application dossier. The CDE review will be provided to the CFDA, after which the CFDA will make its decision on whether to grant approval. Clinical trials must be conducted at approved institutions. Local ECs for the approved institutions are used. This process is conducted in parallel with the CFDA examination of the application and the CDE technical review. The Clinical Trial Management Committee, which oversees clinical trials at the investigational site, must also give approval before a clinical trial can proceed. This should be submitted to the institution’s approving body.
Japan

The Pharmaceuticals and Medical Device Agency (PMDA) offers consultation before clinical trial notification (CTN) submission, to ensure that it meets scientific, data collection, analytical, patient safety and ethical requirements. The approval process is by default, which means that if no objection is received within 30 days, the trial can commence. All documentation must be written in Japanese. To obtain IRB/IEC approval in Japan generally takes 4-8 weeks. Japan also operates a local IRB/IEC process and has a Japanese GCP (JGCP) guideline. This is harmonised with the ICH GCP guideline, although there are considerations specific to J-GCP.
It is preferred that each site uses its own IRB/IEC, but it is possible to get external IRB/IEC approval. As per JGCP requirements the site director would need to ask IRB whether the trial is feasible and the head of each trial location must take responsibility for signing any contracts and overseeing the trial.
New Zealand

The sponsor is required to make an application to the regulatory authority Medsafe and the Health and Disability Ethics Committees (HDECs), seeking permission to conduct a clinical trial and ongoing regulatory reporting procedures in New Zealand (NZ). Medsafe is responsible for the applications and approval process.
NZ has two types of approved EC that conduct ethical reviews of clinical trials: HDECs, which are used for most clinical trial approvals, and the Institutional Ethics Committees (IEC), which are used for late-phase research involving humans, originating from that specific institution. Moreover, Medsafe undertakes scientific assessment of applications to conduct trials and makes recommendations to the Director-General of Health on whether or not they should be approved. NZ has a parallel approval pathway which involves submission to the HDEC and Medsafe simultaneously. Furthermore, NZ does not require Standing Committee on Therapeutic Trials (SCOTT) review for trials involving previously-approved medication. The review and approval timeline is approximately 45 days.
Taiwan
Taiwan uses central and local ethics systems. Multi-site studies use the centralised systems. However, the individual research institutions may choose to accept or reject the Joint Institutional Review Board (JIRB) or Centralised Institutional Review Board (CIRB) decision and replace it with their own.
The centralised procedure aims to reduce repeated local reviews of a study application. For investigational new drug (IND) studies, submission can be done via the JIRB or CIRB.
The JIRB has been governed by the Medical Research Ethical Foundation since 2004. It is gradually being replaced by c-IRB, although some historical studies are still using the JIRB.
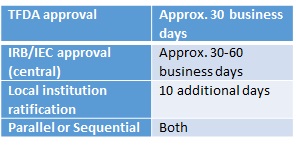
It is important to know that the Taiwan Food and Drug Administration (TFDA) will not grant approval until the Human Research Ethics Committee (HREC) has given its approval. Therefore, it is standard to submit the TFDA package and indicate that the HREC letter will be provided at a later date. Prior to the application to the TFDA the potential research project must be registered in the Taiwan Clinical Trial Network; once registered the application to the TFDA must be made within seven calendar days.
For TFDA approval it takes approximately 30-60 days from the provision of the HREC letter. As well as central approval taking about 30 days, local institution ratifications then take an additional 10 working days and are almost guaranteed. The centralised procedure requires a central application about the proposed trial and submission of data for each site’s IRB requirements. Furthermore, IRB approval can be sought sequentially (before regulatory submission) or in parallel to the regulatory submission. Documentation of IRB approval or submission must be provided in the regulatory package.
Accelerated review may be possible if a trial is multi-country, multi-site research in which Taiwan must be a participant. Parallel submission is also required in any one of these countries: Germany, US, UK, France, Japan, Switzerland, Canada, Australia, Sweden and Belgium.
About the author:
Written by clinical trial organisation George Clinical India.
Read more from George Clinical: