Regulatory consultants: facilitators or obstructors?

There are many benefits to using the services of an external regulatory consultant, who can bring broad experience and help companies gain approvals in a timely manner and with an optimal prescribing label, says Tarun Pandotra.
Pharmaceutical companies face significant challenges in developing new, innovative medicines:
- With only 18 US Food and Drug Administration (FDA) decisions on new drugs expected in 2017, according to BioPharma Catalyst, the number of FDA approvals may continue to decline to a level the industry has not seen since 2007, when 18 new molecular entities (NMEs) and new biologic licence applications (BLAs) were approved.
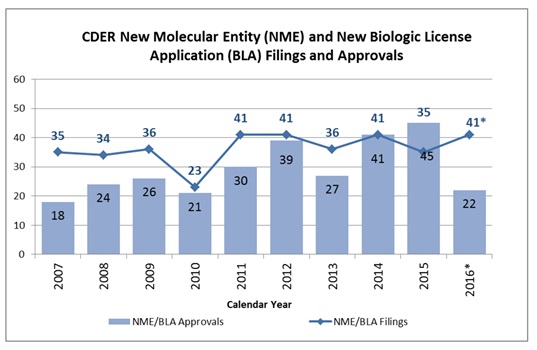
Table 1.
- The failure rate in phase 3 is 40%.
- A Datamonitor study of 346 New Drug Applications (NDAs) found that 42% of submissions received a complete response letter, resulting in a median delay in approval of 13 months.
- Cost cutting and the retirement of baby boomers with deep expertise have resulted in a shortage of in-house experts able to drive innovation and gain regulatory approvals.
External regulatory consultants represent a competent, efficient and unbiased way to address many of these challenges. Regulatory affairs, a key pharma and/or CRO function, where most learning and expertise are gained on the job, is a prominent example. Few universities offer degrees in regulatory affairs, and an academic setting does not provide experience of working directly with regulatory authorities on individual molecules. Actual work on drugs reveals nuanced differences by division, therapeutic area, drug class, and even FDA lead reviewer. This level of understanding is often critical in gaining approvals in a timely manner and with an optimal prescribing label.
Hiring the right regulatory consultant

Table 2 (right) lists the essential skills that a regulatory consultant should have. The challenges of hiring one could be entirely different in the developed world versus developing countries. For reviews and simulations, finding a team which meets needs and works together effectively can be difficult.
For submission work, keep in mind that many consultants in developing countries, like India, Malaysia, Philippines, Sri Lanka, China, Thailand, Vietnam and Indonesia, do not have solid pharma experience and regulatory authority requirements are always changing.
Knowing how to ensure a submission is fileable, reviewable, and approvable requires specific experience, not just extrapolation from early-phase work. In addition, gaining approval for a new indication for a marketed product has some similarities to a new NDA or BLA, but is less complex and has fewer cross-functional challenges.
Clinical trials review teams are often cross-functional, e.g. veteran pharmaceutical professionals from regulatory affairs, legal, medical, pre-clinical, pharmacology, pharmacovigilance, and manufacturing. These consultant teams must be able to work together.
Regulatory outsourcing
Most pharma or CRO companies outsource regulatory tasks to gain capacity or competency. For example, a regulatory affairs chemistry, manufacturing and controls (CMC) department has more work to do than people to do it, or a regulatory authority requires a response about a significant new adverse event in a therapeutic area that is not a strength for the company.
Another approach involves stage reviews and simulations. The beginning and end of each drug development phase involve critical assessments of trial results, plans for future development, plans for meetings with regulatory authorities, preparation of documents,
and, ultimately, submissions. Expert regulatory consultants may help evaluate NDAs and BLAs before filing, prepare for advisory committee meetings, and hold mock advisory committee sessions. This includes identification of gaps, recommendations on improvements, risk assessments, and mitigation strategies. For advisory committees, consultants may help prepare materials, coach clients on how to address issues, and serve as a mock advisory committee panel member. At the actual advisory committee meeting, external experts may coach the client company or participate directly.
Management
Having contracted the right consultant, the challenge is to use them efficiently. Under all outsourcing models, at least one client company contact should be assigned to answer questions about work scope and provide resources such as access to data sets and reports. Usually an in-house subject matter lead is the point of contact. Project management helps ensure the work proceeds on schedule and that the consultants’ time will be used for content work, not caught up in administrative and logistical issues. Face-to-face kick-off meetings and end-of-project meetings are usually worth the expense. Much other work can be done remotely.
Client companies should request feedback on the performance of the consultant from the project managers and in-house subject matter experts frequently and provide it to the consultants or consulting organisation. A rapid halt to the work of any consultant who is not getting the job done is in the best interest of all parties.
Finding seasoned, impartial consultants and setting up effective infrastructure will keep the work focused and productive, and the payoff in on-time approvals with optimal labels is worth it.
Conclusion
As the profile of the regulatory affairs professional continues to grow within the pharma industry, the pressure to provide robust, high quality regulatory submissions steadily increases. The regulatory consultant can be valuable in this process, providing instant access to drug reviews, drug development data, comparative data and regulatory precedents. These can help shape regulatory submissions on a global level by throwing a spotlight on previous submissions within a drug class, specific data requirements and the most successful approval packages.
About the author:
Tarun Pandotra is Director and Founder of Global Regulatory & Consumer Insights. He has 19 years’ experience in drug development. He uses non-conventional development strategies, maximising the use of regulatory mechanisms and innovative approaches to drug development, to bring innovative technologies to patients.
Previously he was a director with PRA Health Sciences, managing clinical operations in South East Asia and India. Prior to that he was a Global Project Manager with PPD. He speaks at national and international conferences.











