Oncology combination drug discovery in the genomics era
Many tumour types find new ways to evade single targeted cancer drugs – hence the push to find new drug combinations to shut down all the escape routes for cancer.
The last 10 years have seen a huge leap forward in our understanding of what causes cancers to develop, known as tumourigenesis.
A number of drivers have been identified, among the best known being BRAF in melanoma and ALK in lung cancer, and we now have a number of novel drugs which target these pathways, delivering unparalleled clinical efficacy in these patient sub-populations. However, there is a downside: although overall response rates can reach 50 per cent or higher, effects are often not sustained. Worse, resistance is possible, and when tumour growth is re-established, it can return in a more aggressive form than in initial treatment.
For this reason, excitement about advances has been tempered with the realisation that these targeted agents may be limited due to tumour cell adaptive responses, or the presence of redundant pro-proliferation or survival pathway signalling. Both types of events appear prevalent, with the latter not particularly surprising given the level of mutation burden observed for the various tumour types characterised to date.
Tumour adaptation
Problematic examples include the RAS/RAF/MEK and PI3K signalling pathways, which are key determinants of tumour cell proliferation/survival and frequently activated in cancer cells. Although there are a few exceptions1,2, in general PI3K inhibitors and MEK inhibitors have not been particularly effective as monotherapies in clinical trials. Pre-clinical studies with both classes of agents highlight the potential for tumour adaptation to drug effects through pathway compensation and/or redundant pro-survival signalling. High residual tumour burden coupled with genomic plasticity and selection can further complicate treatment options due to the potential for generation and selection of (acquired) resistance clones. As a result, there is a need to identify therapeutic solutions to overcome these issues.
The mechanisms by which tumour cells can evade the actions of agents designed to kill them are ill defined and can vary for each drug class and tumour, depending on the collection of genomic events driving proliferation and survival. Single drug activities observed using pre-clinical model systems and results from clinical trials point to the importance of identifying appropriate drug combinations as a critical path for effective treatment of cancer. Combination drugs can address the adaptive responses induced by one or more drugs, as well as suppressing redundant signalling pathways. Oncologists need to identify the multi-target mechanisms that, when impacted, lead to effective and selective synergistic killing of tumour cells.
Good timing
The timing for this type of initiative is ideal. Recent consortium efforts, such as The Cancer Genome Atlas (TCGA) and the Broad Institute's Cancer Cell Line Encyclopaedia (CCLE), are providing detailed genomic information regarding genetic, epigenetic and proteomic alterations that are rigorously associated with particular human tumour types and cancer cell lines3,4. Correlation of genomic signatures between tumours and cancer cell lines will identify those cell lines most similar to human cancer and best suited to serve as models for in vitro combination drug and biomarker discovery efforts. By using cell line models that maintain similar genomic alterations to human cancer, responses to particular combination drugs can be used as an in vitro resource for discovery.
Using cell line models, a key goal for oncologists interested in combination drugs is the identification of drug pairings with more than additive (synergistic) potency shifts or maximal effect increases that ultimately result in tumour cell death (versus cytostatic effects). As an example, consider an oncology drug that has an effect on tumour cell proliferation/survival, where with increasing concentrations of drug, more effect is observed. With combination screens, the aim is to identify a second drug that increases the potency of the first drug or increases overall efficacy (Figure A). The problem is that a priori, one does not know what concentration and ratio of both drugs will result in potent combination effects.
Using a combination high throughput screening (cHTS) strategy, tumour cells can be cultured in multi-well plates and a dose matrix of both agents generated that covers a wide range of concentrations and ratios that facilitate the potential to observe combination effects (Figure B). The assay can be configured such that both cytostatic (Growth Inhibition 100 per cent) and cytotoxic activities (Growth Inhibition 200 per cent) can be distinguished. The rest of the dose matrix consists of various concentrations and ratios of both agents.
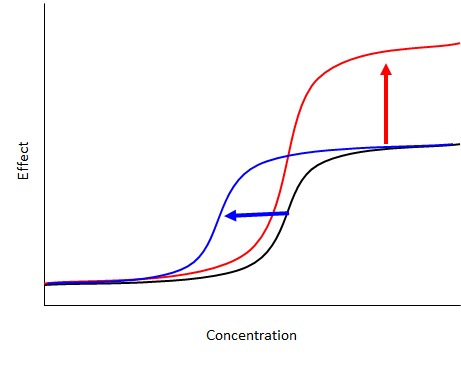
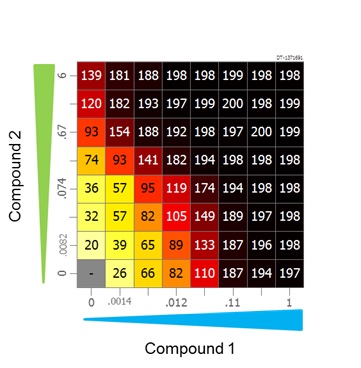
As mentioned, an important consideration for cHTS is that disease-representative tumour models be used, which encompass features found in the patient population. In addition, assays may need to be tailored so that the phenotypic effects of specific single agent or combination activities will manifest. For example, DNA damage response inhibitors and epigenetic modulators are two classes of drugs where multiple cell doublings may be necessary for full effects to be seen, and proliferation rates may vary dramatically for the tumour lines in a screening panel. Additionally, cell-based assays may require the use of specific culture conditions which incorporate disease-relevant microenvironment factors that recapitulate the spectrum or magnitude of growth and survival-relevant signalling pathway activities found in the disease setting.
Evidence for microenvironment effects include changes in drug sensitivities observed when some tumour cells are assayed in commercial media supplemented with growth factors, and changes in drug sensitivities observed when multiple myeloma cells are cultured in the presence of stroma or conditioned media from bone marrow stromal cells5,6. It will be important to recapitulate the complexity of the disease biology as much as possible during the combination drug screen and, if robust screening conditions cannot be defined, such assays (although low throughput) should be considered during follow-up evaluation of primary screen combination leads.
Discovery phase
Combination drug discovery should start early during the drug development process. For many disease-relevant 'critical path' inhibitors, there are already concerns about deployment as monotherapies as early as the discovery phase of research. Cancer disease biology is complex, adaptive and unpredictable, and so it is best to take an agnostic approach and perturb cancer signalling/survival pathways using many drug combinations to identify the conditions that most effectively lead to tumour cell killing. By doing so, we will discover the molecular mechanisms that, when jointly affected, most efficiently and selectively lead to cell death. The evaluation of the potential for combination activity could become a prioritisation step for advancement of a discovery phase lead compound.
Careful pairing of a targeted monotherapy with one or more drugs to improve efficacy or limit a cancer cell's ability to adapt to drug challenge will increase complete response rates, reduce mortality and prolong survival. Capitalising on all available genomic information and resources, including cancer cell line models and patient tumours, predictive biomarker panels tailored to particular drug combinations can be selected. Potentially responsive patient populations where combination drug therapy will be more likely to have a meaningful effect can then be identified, resulting in a dramatic impact on patients' quality of life.
References
1. Gopal, A K et al, 2014. PI3Kδ Inhibition by Idelalisib in Patients with Relapsed Indolent Lymphoma. http://www.nejm.org/doi/full/10.1056/NEJMoa1314583
2. FDA Approves Mekinist, 2013. http://www.drugs.com/newdrugs/fda-approves-mekinist-trametinib-advanced-melanoma-3798.html
3. The Cancer Genome Atlas. http://cancergenome.nih.gov/
4. Barretina, J et al, 2012. The Cancer Cell Line Encyclopaedia enables predictive modelling of anticancer drug sensitivity. http://www.nature.com/nature/journal/v483/n7391/full/nature11003.html
5. Wilson, T R, et al, 2012. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. http://www.ncbi.nlm.nih.gov/pubmed/22763448
6. McMillin D W, et al, 2010. Tumour cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. http://www.ncbi.nlm.nih.gov/pubmed/20228816

About the author:
Dr Richard Rickles joined Horizon CombinatoRx (formerly CombinatoRx and now part of Horizon Discovery) in 2006 and has been responsible for the scientific direction for oncology. His group is engaged in multiple collaborations with pharmaceutical and biotech companies for cell-based assay development and high-throughput screening to discover novel combination therapeutics to treat cancer. Dr Rickles conducted his postdoctoral work at Harvard University in the laboratory of Dr. Mark Ptashne studying the regulation of eukaryotic gene expression. His industry experience started at Ariad Pharmaceuticals, where he worked with Dr Joan Brugge and Dr Mark Zoller on the structure/function of SH3 domains, and development of the first regulated mammalian cell two-hybrid assay for drug discovery. He has also worked at Aventis and Vertex. Prior to joining CombinatoRx, Dr Rickles was in the laboratory of Dr Steve Elledge at Harvard Medical School, working on the development of novel retroviral gene silencing vectors that facilitate the molecular genetic analysis of mammalian cells.
Have your say: Are combination drugs the way forward for treating cancer?









