How does HTA for orphan drugs differ across Europe?
New research looks at the factors that speed up and slow down HTA appraisals for rare disease medicines across Europe.
Rare diseases drugs have always faced challenges when it comes to HTA approvals, even as governments bring in more regulatory policies that make their path through assessment easier.
Several factors make it difficult for HTA bodies often to assess orphan drugs, including a lack of robust trial data due to difficulties in finding patients, the absence of randomised controlled trials, the use of surrogate endpoints, and the lack of active drug comparators.
A new analysis from consulting firm CRA has honed in on the different challenges faced in four European markets – England, France, Germany and Scotland – and looked at how manufacturers can increase the chances of a successful appraisal.
The research analysed more than 70 EMA-approved rare disease therapies and compared reimbursement recommendations from the regulatory bodies in each country, to see how HTA decisions potentially prolonged time to reimbursement.
The results show that HTAs for orphan drugs can vary widely across Europe, causing inconsistencies in evidence requirements and recommendations.
Rates of approval
The study reviewed all 80 European Medicines Agency (EMA) authorised drugs receiving an orphan designation between 1 January 2013 and 31 December 2019, analysing their HTA outcomes and time to reimbursement across France, Germany, England and Scotland.
A comparative analysis was then conducted on the 71 approved drugs that achieved a negotiated price in at least one of the four markets.
Germany had the highest approval rate of orphan drugs at 98% – however most of these recommendations (73%) were awarded a ‘non-quantifiable benefit’ rating, the automatic rating for an orphan drug, which shows the regulator did not see any benefit compared to comparator products (see graph 1). The authors also note that orphan drug trials with higher p values and surrogate endpoints are often accepted for assessment in the country.
A more favourable outcome from the German regulator took on average 1.4 times longer to achieve a final negotiated price (708 versus 510 days).
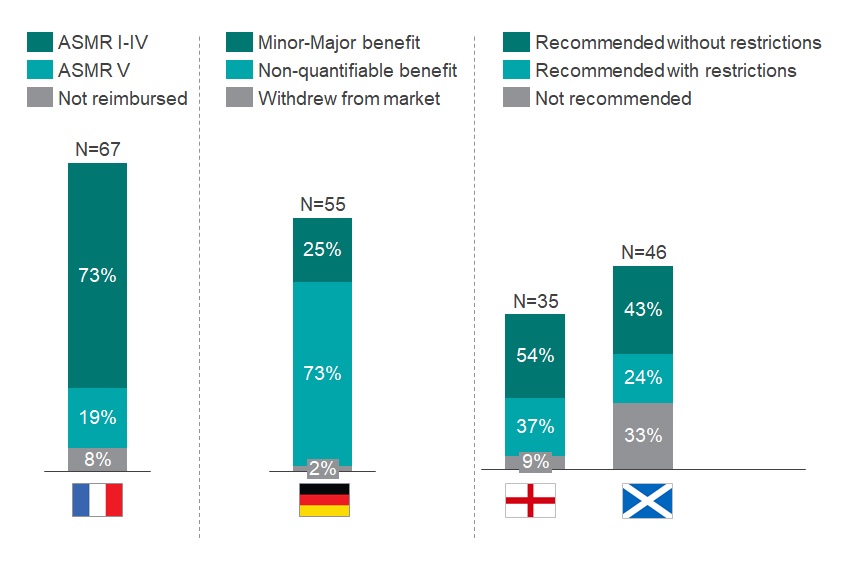
Graph 1: Assessment of the HTA outcome in France, Germany, England and Scotland of all orphan drugs that obtained an EMA approval between 2013-2019. N above each bar equals the number of drugs reviewed by the respective HTA body. Source: CRA Analysis
France and England had comparable approval rates (92% and 91%, respectively); however, France reviewed almost twice the number of orphan drugs over the period of analysis (67 versus 35). Only 19% of the orphan drugs in France were awarded an Amelioration du Service Médical Rendu (ASMR) rating of V, which indicates no improvement in medical benefit. Drugs with an ASMR IV-V rating were reimbursed in 427 days, compared to 585 days for products with ASMR I-III (see Graph 2).
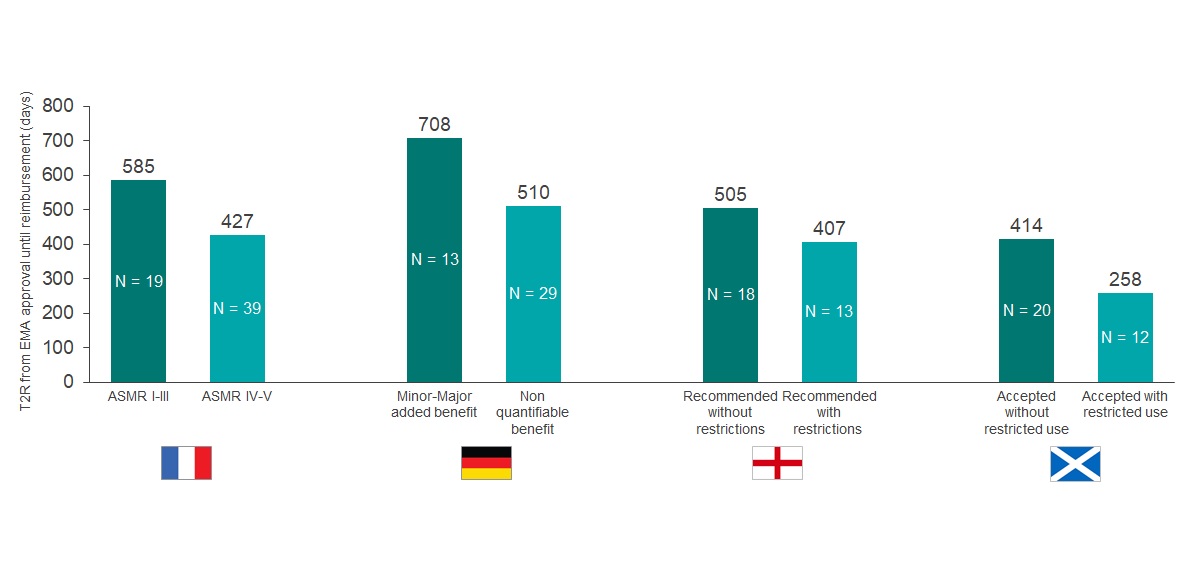
Graph 2: Comparison of the time to reimbursement (days) for EMA orphan drugs approved from 2013- 2019. Orphan drugs were reimbursed in at least one of the selected markets (N = 70). N within each bar equals the number of drugs with each outcome reviewed by the respective HTA body. Source: CRA Analysis
In England, two key mechanisms were often used to achieve approval: label restrictions or a patient access scheme (PAS).
Over one third (37%) of orphan drugs appraised by NICE only achieved approval in a positioning or population that was restricted versus the full regulatory approved label. Companies that accepted such restrictions saw faster approval time compared to no drugs with restrictions (407 versus 505 days).
Meanwhile, although introducing a PAS improved the chance of approval, the analysis suggests that it actually delays the overall appraisal time (523 versus 311 days).
Scotland had the highest rate of non-approval for orphan drugs. Thirty-three percent of drugs reviewed were not accepted, despite specific modifiers in place for rare disease products, including the incorporation of the patient voice through the country’s Patient and Clinician Engagement (PACE) meetings – which were included in 74% of orphan drug submissions between 2013-2019.
Improving HTA outcomes
The authors conclude that while the various concessions and modifiers introduced by different governments have a positive impact on minimising rejections and accelerating approval times, there are still challenges in capturing the full value of orphan drugs within the HTA process.
“Achieving more favourable outcome ratings, avoiding restrictions, or addressing uncertainty with a PAS all lead to prolonged appraisal times,” the authors say. “Manufacturers are therefore still required to consider carefully their HTA launch strategy and complement this with additional evidence generation and engagement from a wider stakeholder group.”
The authors outline several approaches companies can take to improve the chances of success in orphan drug approvals and overcome the challenge of having limited data and evidence available.
One approach is to agree methods for ongoing real world data collection post-launch with HTA bodies.
They add that “creative” solutions to real world evidence collection could help, such as developing apps for patients and HCPs.
Meanwhile, it is also important to strive for wider engagement with the rare disease community and other stakeholders.
Listening to views from patients and HCPs can help with processes like Scotland’s PACE meetings – but more indirect forms of stakeholder engagement may also improve HTA outcomes, as the value-added services provided to these stakeholders can be leveraged during negotiations.
“For example, Galafold, an enzyme replacement therapy for Fabry’s disease, is primarily differentiated from existing treatments by providing a reduction in administrative burden,” the authors say. “Despite this, Galafold was able to achieve an ASMR IV in France, recommendation by NICE, and was accepted for restricted use in Scotland.”
They note that the perception of Galafold’s value may have been improved by the additional value-added services the manufacturer, Amicus Therapeutics, offered to a wider stakeholder group
For example, Amicus reimbursed amenability tests for patients with unknown mutations that could be referenced against Galafold’s amenability table via a physician support website. This service was accepted in the NICE evaluation as something which avoided additional resource implications for the NHS.
For more information contact the report’s authors:
Steven Kelly – skelly@crai.com
Ioanna Stefani – istefani@crai.com
Charlotte Poon – cpoon@crai.com
Nimisha RaJ – nraj@crai.com











