Who needs (early) advice? You do!
With increasing emphasis on real-world evidence in the drug approval process, Don Husereau examines the current status of, and future prospects for, Health Technology Assessment Early Scientific Advice programmes.
Different evidentiary requirements and decisions taken by regulators and Health Technology Assessment (HTA) bodies can prove a hard lesson for some, who have provided evidence that their drug can work, but not enough to convince payers it will work in the real world. This has led to a demand for advice on trial design, outcomes and other important pieces of information that will satisfy payers as well as regulators. While regulators have offered advice on evidentiary requirements for years, HTA bodies have started doing this only recently. However, these systems of HTA Early Scientific Advice (ESA) and Dialogue are evolving quickly.
Forays into early advice
ESA processes with manufacturers, often undertaken jointly with regulators, have typically been brokered by HTA and regulatory bodies and, in some cases, via private sector consultancies or academic initiatives. They have been welcomed by manufacturers, some of which have merged outcomes research and market access processes and departments into developing entities more fully focused on HTA.
While some jurisdictional programmes have become formalised, even operating at arm’s length from, or independently of, the HTA bodies themselves, others have been less formal (e.g. Spain), or pilots. For example, in 2009 Australia piloted a process that brought industry together with the HTA body and the regulator to focus on matters of ‘common interest’. It appears to have “provided early and clear signals to inform major development investments and the probability of successful market access”.
Table 1: Early scientific advice processes internationally
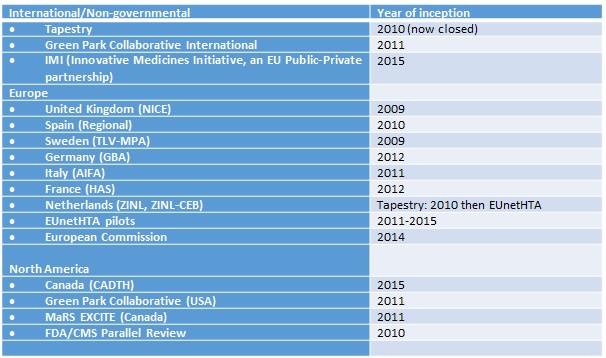
A greater effort to provide ESA emerged from the activities of the European Commission(EC)-initiated European network of HTA bodies (EUnetHTA). Specifically, joint action work through this project led to ESA pilots and prompted a similar EC collaborative project, called the Shaping European Early Dialogues (SEED) initiative that built on these efforts. In support of this work, the European Medicines Agency (EMA) had already begun to invite HTA bodies to its early advice meetings, called 'parallel' advice, with the aim of modernising regulation through collaboratively designing clinical development programmes, called adaptive pathways. Combined, these projects resulted in dozens of consultations by the end of 2015 (63 EMA parallel advice procedures, 19 EUnetHTA consultations) and the development of consensus ‘best practices’ in the European approach to ESA.
ESA programmes today
Experience with these schemes has led to some similarities in approach across ESA programmes, including the need for preliminary agreement on the questions asked, standardised reporting approaches to describing the technology and payer decision problems through the use of briefing books, and the use of face-to-face meetings.
However, subtle, but not insignificant, differences have emerged too. These include the ability to include patient input, the nature of the face-to-face meeting (consultative versus a dialogue), flexibility in scope of questions, timing in the phase of clinical development, and what is delivered to companies.
There has also been recognition of the value of joint advice either across HTA bodies or with regulators. The latter approach means companies do not have to try to internally coordinate meetings with HTA bodies and regulators independently. It also has the distinct advantage of being able to clarify or better manage advice from these two entities that may seem conflicting. Based on the success of the pilot programme launched by EUnetHTA, a formal multi-HTA advice programme is anticipated.
Companies may also want to consider seeking input from outside Europe, where there is much ESA activity. Initiatives like the Green Park Collaborative in the US or the CADTH Scientific Advice Programme in Canada may give them a wider perspective on clinical development programmes using well-established HTA expertise.
What advice? When and where ESA is useful
Some companies may find navigating this ESA patchwork difficult. They can now receive advice from HTA bodies individually through formal or informal jurisdiction-specific processes, through an HTA body’s participation in EMA consultations, or through the developing EUnetHTA programme. Certainly some schemes have the advantage of involving multiple HTA bodies and parallel advice from regulators that may be desirable. There are also differences in timing and the types of questions that can be asked.
Companies should consider how much alignment there is between HTA and regulatory thinking internally, whether they are able to act on the advice, and the potential for the advice to de-risk their investments. Some examples of these are shown in Table 2.
Table 2 Examples of early advice/dialogue programmes for pharmaceutical innovators
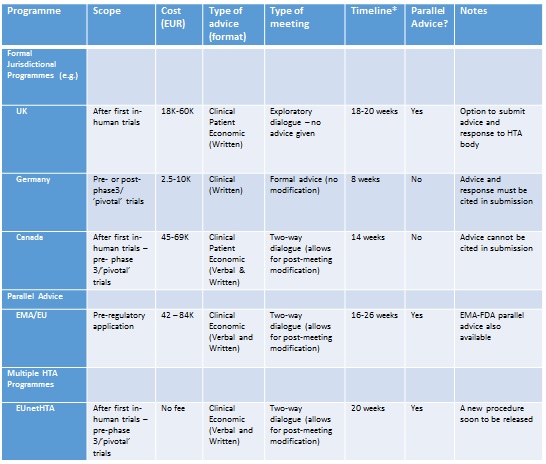
*Time from Briefing Book submission to delivery of advice.
Evolving paradigms: what next?
The soon-to-be-announced multiple HTA advice procedure, under EUnetHTA’s joint action 3, should provide an improved process, based on previous pilot and SEED experiences. This may further respond to industry needs for consolidation or mutual recognition of advice across jurisdictions. It may also bring flexibility in scope and timelines when seeking advice. Certainly, the option to seek parallel regulatory advice is still seen as valuable, and some programmes without this option (such as Canada) are investigating the possibility.
ESA may also be the next step towards a more holistic approach to creating adaptive pathways. Some practical questions, such as who pays and who participates, may still need to be answered as programmes continue to evolve. Given the apparent enthusiasm in Europe for this type of advice, it is sure to become integral to managing risks in clinical development in the future. Participation may also help companies demonstrate value more consistently, and increase their strategic advantage.
About the author:
Don Husereau is an Adjunct Professor of Medicine at The University of Ottawa, and a Senior Associate with the Institute of Health Economics. His research focuses on appropriate and innovative approaches to the use of evidence and economics to inform health policy.
He is chair of an International Task Force that developed consolidated health economic evaluation reporting standards (CHEERS) and served as Scientific Secretary for the HTAi Policy Forum, which explored HTA ESA in 2015-16. He serves on the pan-Canadian Oncology Drug Review Expert Review Committee (pERC) and Ontario Committee to Evaluate Drugs.
Read more on market access:









