The EMA’s Adaptive Pathways: are safety concerns exaggerated?

The EMA’s Adaptive Pathways initiative represents an opportunity for drugs in areas of high unmet need – but companies still need to provide a compelling case for efficacy, says Janice Haigh.
The European Medicines Agency’s (EMA) innovative ‘adaptive pathways’ (AP) approach – an effort to improve timely patient access to new medicines in areas of high unmet need – allows for early and progressive patient access to a medicine using the existing EU regulatory framework.
Products accepted into this approach are granted conditional marketing authorisation based on early data. Patients are recruited into observational studies and these studies support the subsequent full marketing authorisation.
The AP approach is based on the principles of iterative development throughout the lifecycle of a medicine, where clinical trial data is supplemented by real-world evidence.[i] A key principle is early patient and health technology assessment body engagement. AP seeks to achieve an appropriate trade-off between unmet need and a wider-than-traditional degree of uncertainty around risks and benefits. This approach potentially expedites access to new medicines, while at the same time ensuring that the evidence gap is progressively filled.
The decision on when to grant marketing approval for a drug reflects a balance between the desire for patients to have access to promising new medicines as soon as possible, while still ensuring that these medicines are as safe as possible (Figure 1).
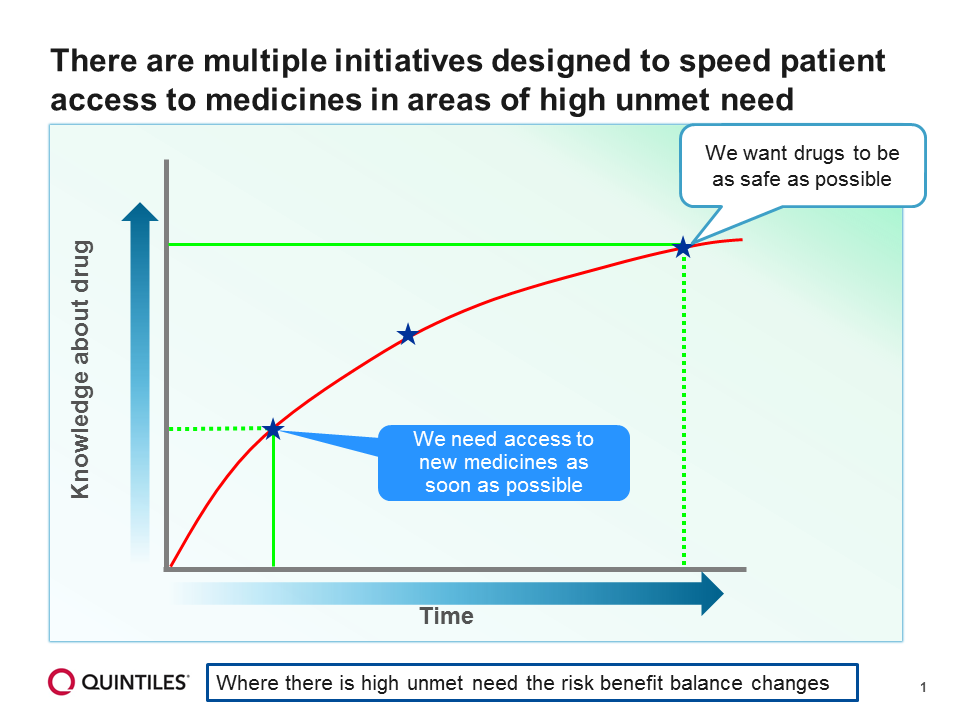
Figure 1: Speeding up patient access to medicines in areas of high unmet need
For patient populations with a life-threatening disease and no existing treatments, tolerance for risk may be higher, with the potential to allow access to a new drug while collecting additional data. The AP approach is intended to provide timely access to new medicines by balancing medical need with evolving information on the benefit-risk (Figure 2).
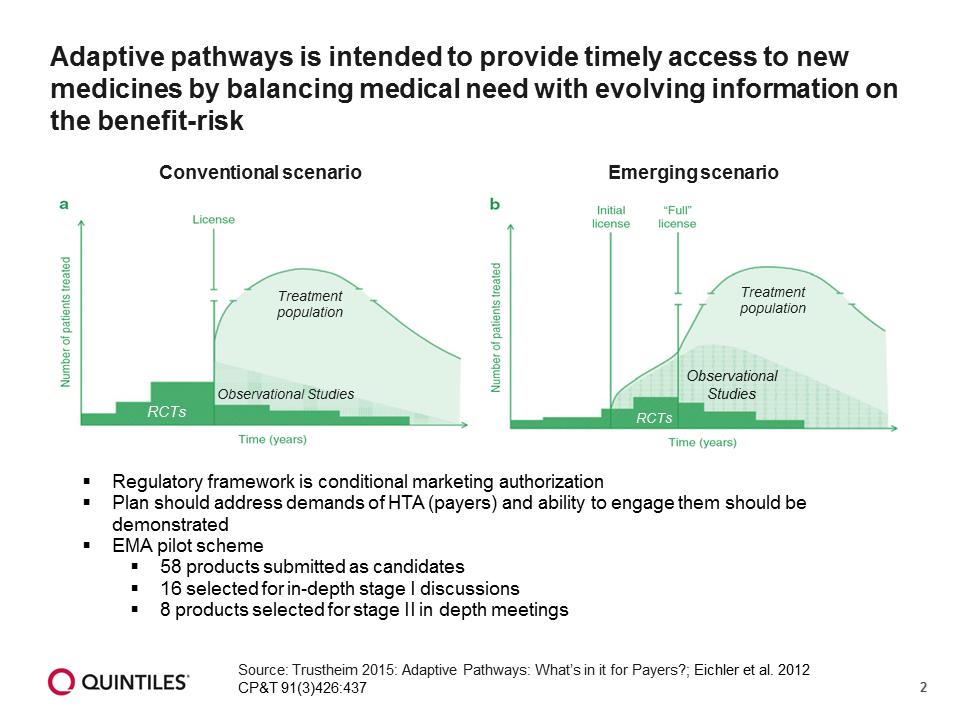
Figure 2: Balancing medical need with evolving data on benefit-risk[ii]
Safety concerns appear to be exaggerated
The AP initiative has faced criticism for its perceived potential to lower safety standards by allowing new products onto the market more quickly.[iii] This criticism tends to assume that the role of the regulator is to guarantee safety; it is not. The role of the regulator is to assess risk-benefit. In practice, most safety issues are observed after marketing authorisation, when large numbers of patients have been treated. Efficacy is the most frequent reason for failures in late-stage development (Figure 3).
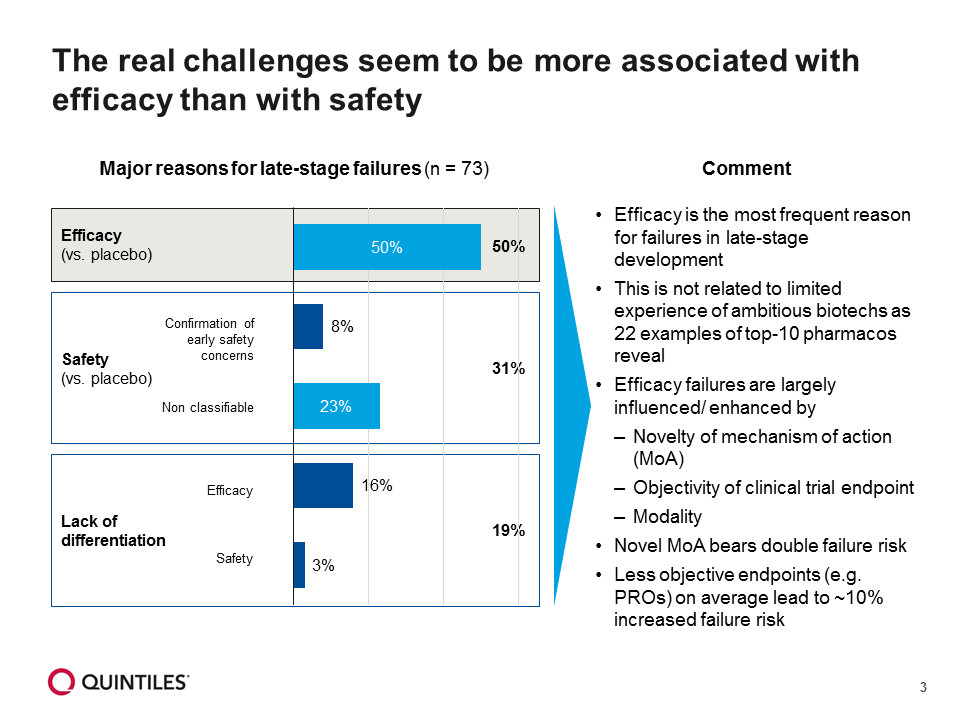
Figure 3: Major reasons for late-stage failures
The real challenge to the AP concept is more likely the practical one associated with measuring efficacy or effectiveness in an observational study design. The issues are probably disease- or product-specific, and will involve issues such as randomisation, dosing, choice of comparator, recruitment, a complicated disease pathway or patient population, or the fact that multiple centres for rare diseases may use different approaches and standards of care.
HTA will likely be challenging
Health Technology Assessment (HTA) bodies have not yet evaluated any product accepted for AP, but there are analogous situations. The most obvious one involves products given conditional marketing authorisation (CMA) by the EMA, as this is the regulatory route for AP. As a result, the products share the situation that: the benefit-risk balance needs to be positive; no changes are made to the standard for evaluation; the benefit of immediate availability of the product to the patient must outweigh the risks related to incomplete data; and there is a need for continued evidence gathering with comprehensive data required after marketing authorisation application (MAA).
A second situation involves extensions of indications. Going from initial indication to full indication in the AP is analogous to an extension of indication from a payer’s perspective (e.g. extending to a different disease stage or different treatment line/stage).
A third involves clinical evidence based on single arm trials, which share with products accepted for AP the feature that only limited clinical evidence is available.
To date, 17 products have received a CMA, of which 10 were orphan and 12 were for oncology indications. Evidence suggests that drugs with a CMA have a higher probability of receiving a negative recommendation than the average of all HTAs, based on data from Quintiles HTA Accelerator[iv] (Figure 4). However, unmet need does seem to be taken into account.
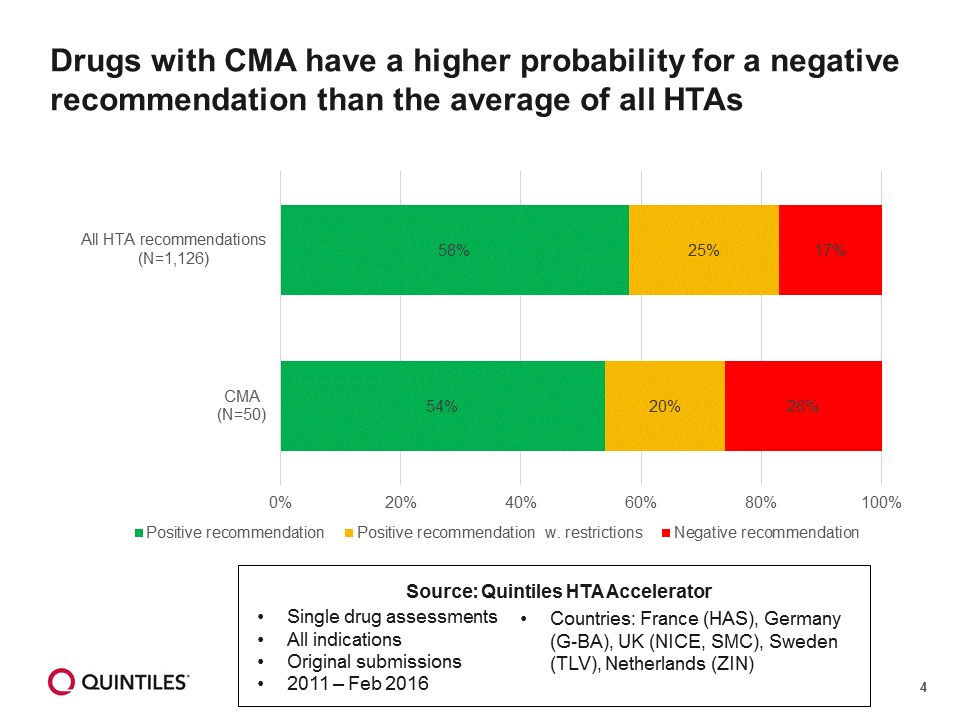
Figure 4: HTA outcomes for drugs with conditional marketing approval
Compared with other HTA bodies, the French HTA has typically given higher ratings for drugs with CMA (Figure 5).
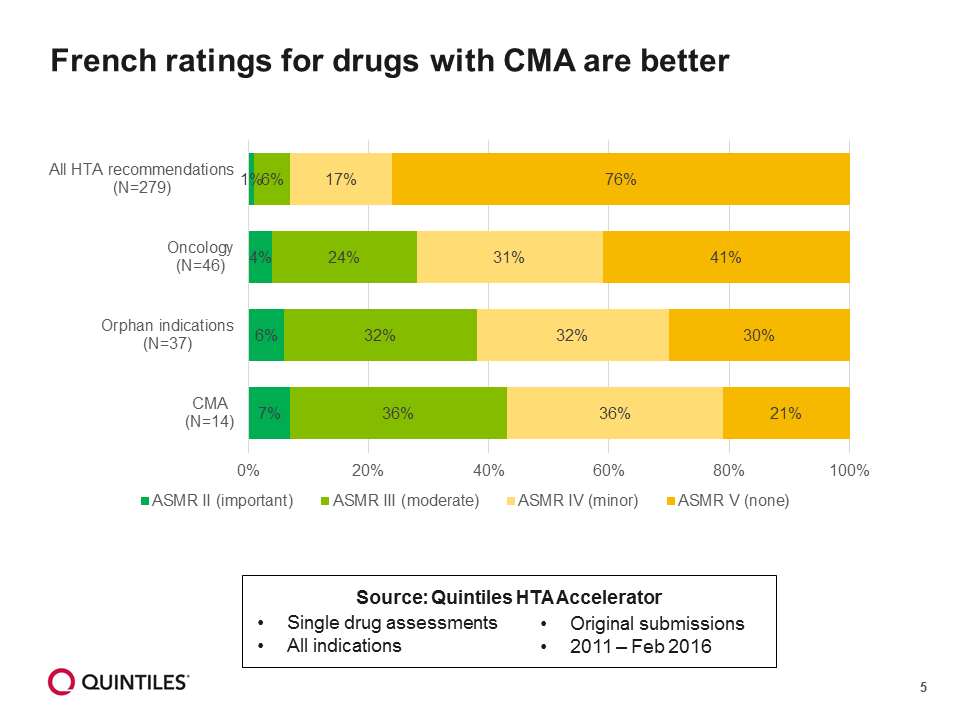
Figure 5: French ratings for drugs with CMA
The time from regulatory approval to HTA recommendation is, however, shorter for drugs with EMA conditional approval (Figure 6).
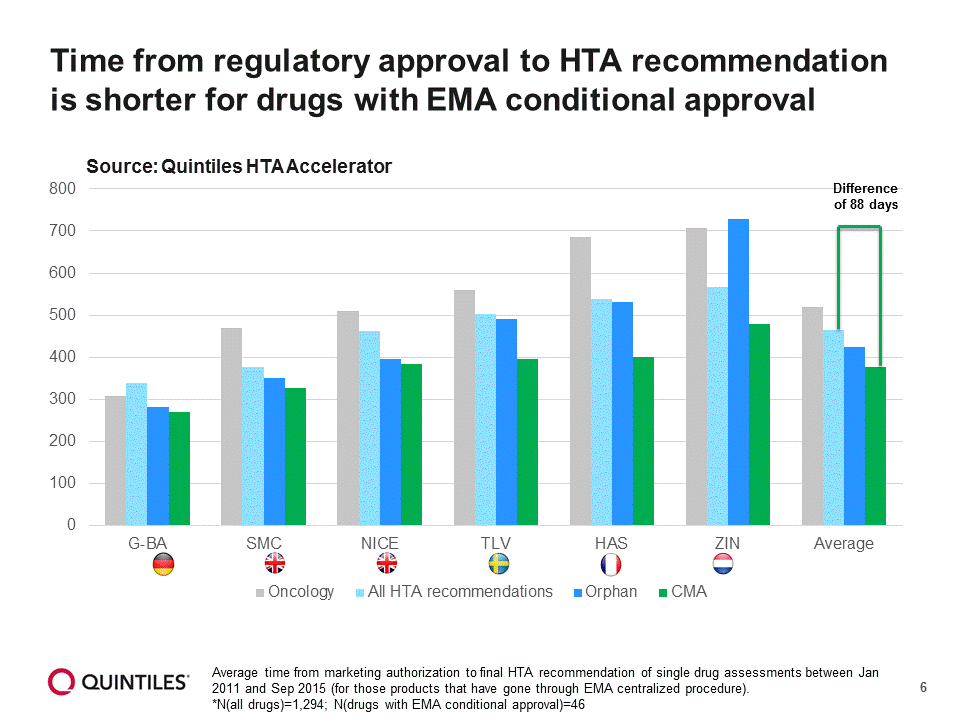
Figure 6: Time from approval to HTA recommendation
Even with all the essential elements in place, some products will fail. Critical success factors include a credible development plan, controlled access, management of risks and strict surveillance.
Key challenges for AP and learnings from analogous situations include:
- Relatively many rejections for HTAs for drugs with EMA conditional approval and extension of indication indicate that these assessments will be challenging for products accepted for AP
- Limited clinical data might be an issue, since clinical benefit was a key decision driver for drugs with EMA conditional approval. In addition, payers have highlighted the uncertainty of results in clinical and cost-effectiveness due to immature data, and in some cases, this uncertainty has resulted in negative recommendations by payers. As a result, it is essential to prepare for different interpretations of the same data
- Emphasising the unmet need will be key.
Companies can improve their chances of positive HTA by undertaking effective consultation with Health Technology Assessment Bodies (HTABs; Figure 7). Acceptance onto the AP pilot requires companies to undertake parallel scientific advice. This is one environment where closed questions are more powerful than open questions, and the quality of advice depends entirely on the quality of questions asked. HTABs cannot tell sponsors what to do, and are only able to respond to specific questions. It is therefore essential to support questions and concepts with data and analysis. For HTABs that use cost-effectiveness analysis, developing an early health economic model allows for highly specific feedback. In addition, the EMA and HTABs will need to see solid plans for future development and analysis.
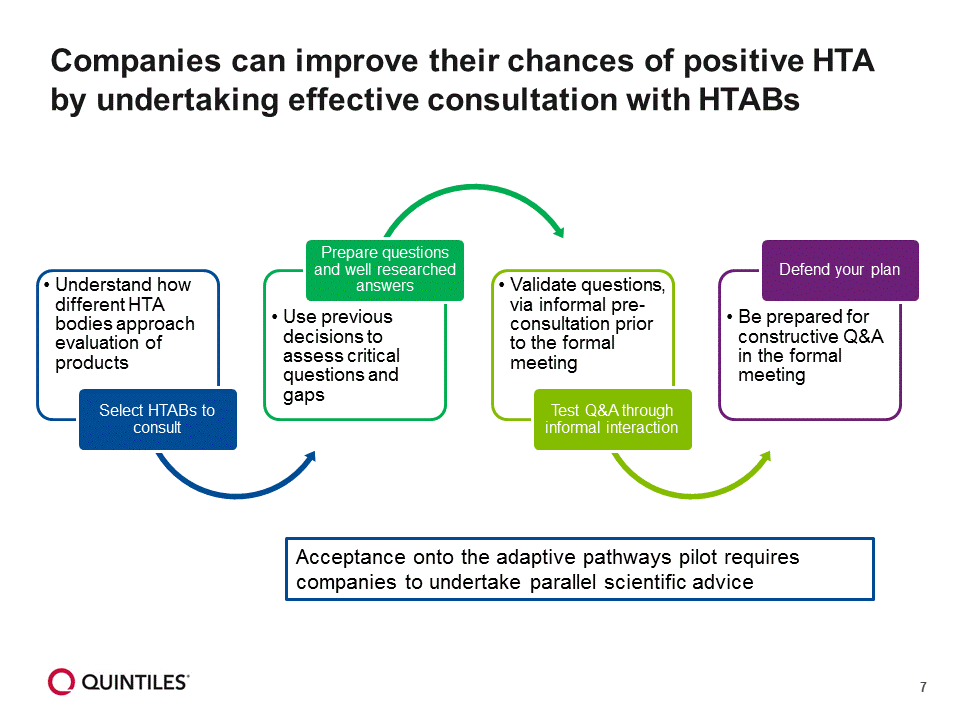
Figure 7: Effective consultation with HTABs can improve the chances of positive HTAs
Conclusion
The most appropriate therapy to take through AP is one with a significant unmet medical need. Safety concerns are not typically an issue with this regulatory approach, since most safety issues do not arise until after marketing authorisation, when large numbers of patients have been exposed to the therapy.
The role of the regulator is to determine that there is a positive risk/benefit balance, rather than to seek absolute proof of safety. In contrast, the challenge of demonstrating efficacy or effectiveness may have been underestimated. It is especially hard to prove efficacy without randomisation, which is a luxury not available in real-world evidence.
The AP initiative is an ambitious attempt by EU regulators to provide patients with earlier access to medicines. The future of this programme is unsure, although a regulatory effort to help meet unmet medical needs will most likely continue. In conclusion, the AP approach is still at its beginning, with the final destination far from certain.
[i]http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000601.jsp
[ii] Eichler, H. G., Oye, K., Baird, L. G., Abadie, E., Brown, J., L Drum, C., ... Hirsch, G. (2012). Adaptive licensing: Taking the next step in the evolution of drug approval. Clinical Pharmacology and Therapeutics, 91(3), 426-437. 10.1038/clpt.2011.345
[iii] Trusheim M. Adaptive Pathways: What’s in it for Payers? Presentation at the 20th ISPOR Annual International Meeting on May 19, 2015.
[iv] http://www.quintiles.com/landing-pages/hta-accelerator
About the author
Janice Haigh is Principal, Advisory Services, Quintiles









