Are we dismissing the ‘business’ of clinical research?
Many of us concur that the biopharmaceutical industry is facing a crisis, and that the R&D process needs to transform entirely in order to address skyrocketing costs and minimal effectiveness. While we are starting to see advancements in scientific technologies that address the challenges with early phase research, discussions surrounding the operational and financial aspects of clinical trials do not seem to be addressed frequently. This article will discuss these differences, and address simple and effective ways of reducing R&D costs through effective business models in clinical research.
The rut the biopharmaceutical industry is in
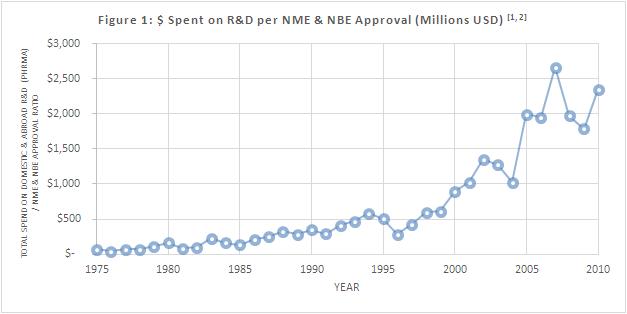
The biopharmaceutical industry is experiencing significant challenges surrounding cost control and strategies in R&D. Figure 1 demonstrates that the amount of capital spent on R&D has continually increased since the 1970s, and has become increasingly volatile since 2004 1, 2. In 2010, the cost of receiving an NME/NBE was $2.35 billion.
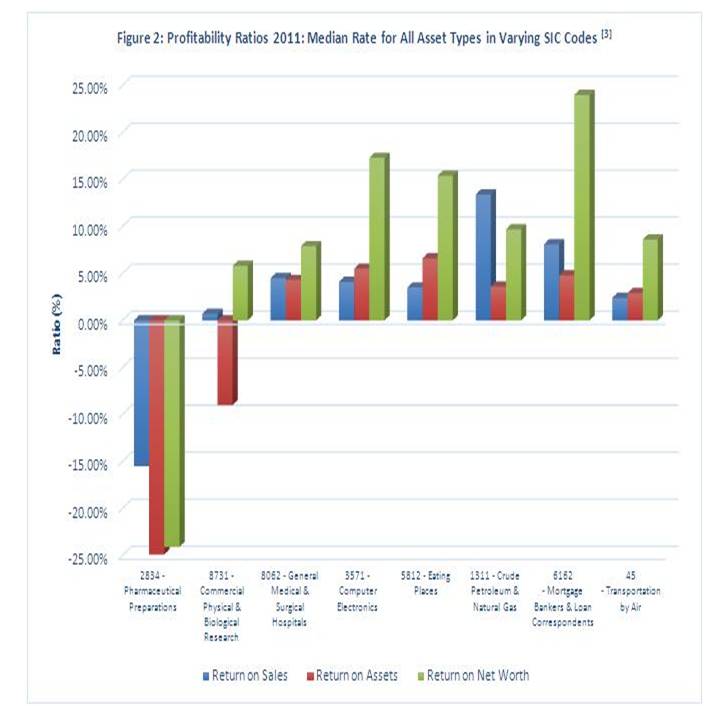
Furthermore, the biopharmaceutical industry continues to experience sluggish profitability. Figure 2 illustrates a variety of financial ratios for varying SIC codes, and the biopharmaceutical sector (SIC 2834) exhibits negative median profitability compared to other industries 3. While some biopharmaceutical enterprises are highly profitable, these negative outcomes make sense due to the high failure rate of medical innovation (particularly with small biopharmaceutical enterprises), as five out of every 5000 innovative compounds will reach clinical trials, and only one will receive FDA approval 4.
Forbes recently released a video that involved R&D executives from biopharmaceutical enterprises to discuss the challenges of R&D. During this discussion, there was a lot of talk around improving the R&D process through scientific and medical productivity. For instance, Sean Harper of Amgen indicated that the challenges of R&D productivity involves targets (i.e. time / money / opportunity costs), and that biomarker / genetic research enables human and biological validations of preclinical research rather than utilizing animal models, which would, subsequently, improve R&D productivity.
Some enterprises, such as WuXi PharmaTech, which specializes in offering a gamut of laboratory solutions in the drug discovery process (including genomics), is capitalizing on the need for cost efficiency during drug discovery, as it has grown its annual net revenue base from $253.5 million – $499.9 million from 2008–2012, respectively. We are also starting to see the emergence of analytical technologies, such as Quantitative Medicine, which leverages data in order to predict a medical product's outcome on pharmacokinetics and failure rates, and enables molecular optimization prior to conducting animal and human studies; in essence, these technologies could reduce the failure rates during clinical trials through preclinical optimization.
Despite breakthroughs in scientific innovations, there seems to be minimal talk about the business value of enhancing productivity in clinical trials.
Simple and effective business models for cutting R&D costs
According to a report from The President's Council of Advisors on Science and Technology, high costs and sluggish productivity associated with R&D involve not only long time to market, regulatory uncertainty, and high failure probabilities, but particularly high expenses in the drug development process, with clinical trials being the most expensive 1. Many of the aspects affecting clinical trial productivity are business in nature.
A combination of suboptimal protocol design, and substantial increases in data collection are placing financial and time strains on not only sponsors, but also study sites and subjects. To demonstrate, mean endpoints / protocol increased by ~100%, and the total number of procedures increased by 57% from 2000–2010 5. Moreover, complex inclusion and exclusion criteria attributed towards clinical trial subject recruitment delays and retention difficulties 5, 6.
Suboptimal protocol design also increases clinical trial time and costs due to protocol amendments. The average number of protocol amendments for phase I, II, and III trials were 1.9, 2.7, and 3.5, respectively, and the costs associated with each protocol amendment was estimated at ~$454,000 (excluding internal resources, re-submissions to local authorities, and translation fees) 7. Changes in study strategy, protocol design flaw, recruitment difficulty, and inconsistency / errors in protocols ranked in the top 10 causes that triggered protocol amendments. It is of particular interest in this context that the first amendment is in the majority of the cases implemented before the first patient was included into the trial 7.
Implementing quality by design techniques via protocol optimization offers many benefits. Optimizing protocols to focus on specific data points that are critical towards achieving IND approvals and post-marketing outcomes cuts down on unnecessary data collection during the trial, which would, correspondingly, reduce monitoring, and patient visits. Empirical research has shown that optimizing protocols by cutting down on the number of CRF pages collected could reduce operational costs by over 40% 9. Furthermore, leveraging analytical techniques to gauge protocol enrollment criteria with existing patient populations, can improve protocol inclusion/exclusion criteria compatibility with actual patient populations, and optimizing site selection strategies through analytical techniques can act as a natural catalyst towards enhancing subject enrollment rates.
Another area of concern to the biopharmaceutical industry involves FDA's recent guidance on risk-based monitoring, which was designed to address the high costs associated with monitoring activities. Monitoring activities represent well over 50% of a clinical trial's budget 8, and current monitoring practices, involving 100% source document verification, has a minimal impact on uncovering trends affecting critical data points 6. According to FDA's guidance, centralized monitoring practices are meant to enhance the detection of unusual and fraudulent trends through statistical analyses, and enable sponsors to focus on critical data points affecting patient safety and data integrity associated with IND approvals.
While Transcelerate has issued a position paper on risk-based monitoring, much of the industry continues to exhibit a level of bewilderment on the topic, as clinical research personnel are not fully equipped with the necessary skill sets required to conduct centralized and statistical monitoring, which requires data analysis, and risk interpretation techniques. Moreover, Transcelerate's methodology on risk-based monitoring did not seem to meet inspectors' expectations (particularly in Europe), as the guidance document does not provide data proven rationale for some of the steps described. Further, risk-based monitoring ties closely with developing quality management systems, such as evaluating IT system infrastructures with protocols to identify analytical parameters for risk-based monitoring plan development and execution.
Nonetheless, the benefits of risk-based monitoring are tremendous, as implementing such techniques can allow sponsors to expand into conducting clinical trials in developing nations without having to risk submitting clinical trial data that may contain fraudulent and low data quality.
The gaps in post-marketing communications and R&D
Other factors affecting protocol design and amendments include changes in post-marketing strategies for clinical trial data collection, given healthcare reform and subsequent pressures affecting pricing strategies. Biopharmaceutical enterprises are now implementing post-marketing strategies to demonstrate the value of their medical products, which includes quality of life, and comparative effectiveness measures 10, 11.
Despite these efforts, biopharmaceutical enterprises are unable to implement communications campaigns post IND approval that fully leverage data that could have been collected during clinical trials in order to demonstrate the value of their medical products. For example, sponsors can conduct analytical and statistical validations of patient safety and medical effectiveness outcomes on QOL between the novel product and a comparator to demonstrate the value of their product to payers and prescribing physicians; doing this in a seamless manner requires planning analytical factors in questionnaire design and clinical trial data collection with post-approval marketing objectives.
Unfortunately, the gap currently exists with internal communications between marketing and R&D departments during the protocol development phase due to natural concerns stemming from compliance and regulatory violations with unapproved investigational product promotion during clinical trials. However, with proper guidance, incorporating communications expertise during the protocol optimization stage (particularly during Phase III trial setup) can enhance data collection strategies to ameliorate post-marketing directives.
How soon will we see advancements in R&D productivity?
While it is unlikely that we will reach the rates of R&D spend / NME approval in 1975 (Figure 1), it is probable that we would realize improvements in productivity to cut the aforementioned rate by 50%, and possibly more. Breakthrough scientific technologies during the discovery phase will likely reduce the failure rate of new medical products, and correspondingly, reduce R&D costs; this would, naturally, improve biopharmaceutical ROI in the long run. Additionally, changes in regulatory approval pathways to improve FDA communications efficiency with biopharmaceutical sponsors would also enhance NME approval rates. Nevertheless, business innovation in clinical trials can result in tangible and immediate financial improvements in R&D. So, why not focus on the business of clinical research?
References
1. http://www.whitehouse.gov/sites/default/files/microsites/ostp/pcast-fda-final.pdf
2. http://keionline.org/sites/default/files/phrma_profile_2011_final.pdf
4. Abbott, Ryan. Big Data and Pharmacovigilance: Using Health Information Exchanges to Revolutionize Drug Safety. Abstract Iowa Law Review 04/30/13
5. Kenneth Getz, Et Al., New Governance Mechanisms to Optimize Protocol Design, Therapeutic Innovation & Regulatory Science 10 July 2013
6. TransCelerate's Position Paper on Risk-Based Monitoring
7. Kenneth Getz, et al. Measuring the Incidence, Causes, and Repercussions of Protocol Amendments. Drug Information Journal, May 2011 45:265
8. Robert M Califf. Clinical trials bureaucracy: unintended consequences of well-intentioned policy. Clin Trials 2006 3:496
9. Eric Einstein et. Al. Sensible approaches for reducing clinical trial costs. Clin Trials 2008 5: 75
11. http://www.bloomberg.com/news/2013-07-11/celgene-says-revlimid-met-study-goal-in-new-myeloma.html

About the author:
Moe Alsumidaie is the President & Chief Scientific Officer of Annex Clinical, a biopharmaceuticals business analytics consulting firm. Moe Alsumidaie can be reached here. Join the discussion at the Breakthrough Solutions in Clinical Trials & Healthcare Group.
Closing thought: How soon will we see advancements in R&D productivity?









