Part Two: Navigating the complexities of global market access for rare and complex conditions

The journey to global market access for innovative therapies is fraught with challenges that vary significantly across the world. Drug manufacturers must adeptly navigate ever-changing regulatory frameworks, cultural expectations, and commercial landscapes to maximise the potential of novel treatments.
This complexity is especially pronounced in the realm of rare diseases, where the emphasis on cost-effectiveness and value can differ widely from one market to another. As decision-makers prioritise different criteria for reimbursement, understanding the intricacies of these varied landscapes becomes essential for manufacturers seeking to ensure that therapies reach the patients who need them most.
Challenges in global market access
At the heart of global market access lie the strategies and processes that drug manufacturers use to navigate the complex regulatory, cultural, and commercial landscapes unique to each target country or market. By mastering the art and science of global market access, companies can optimise the commercial potential for novel therapies, open new opportunities, enhance engagement with diverse rare disease communities and leverage the vast potential available in various global markets.
The emphasis on cost-effectiveness also differs from one region to the next. One study assessing the importance of criteria to decision makers in multiple countries has quantified some of these differences, showing that criteria, such as cost-effectiveness, may be valued extremely high in some jurisdictions, but deemed irrelevant in others.
Within the EU, each Member State is individually responsible for managing its own healthcare system, and there is great variation in market access conditions, local pricing, and reimbursement policies. Consequently, certain products may be accessible to patients in some Member States, but not others, based upon their perceived value and impact on health budgets. As seen in the introduction of CAR T-cell therapies, some therapies are considered unaffordable or not cost-effective, either because product efficacy is unproven or because the established value does not justify the price tag.
Global pricing and demonstration of value for rare disease therapies
To determine the perceived value for money a new drug delivers compared to the cost of other available interventions, decision-makers rely upon incremental cost-effectiveness ratios (ICERs) that contrast newer medicines to the current standard of care. The ratio, known as quality adjusted life year (QALY), is a measure of how well medical treatments improve or lengthen patients' lives.
QALY has been the standard measure in economic evaluations and is used widely in many countries where the HTA guides decision making, enabling payers and regulators to benchmark new products to the QALY threshold. If a drug falls under a designated threshold, it theoretically provides a cost-effective use of financial resources vs the available treatments that it may displace. While the cost-effectiveness threshold varies by country, it is often expressed as the upper limit of a payer’s readiness to pay for a perceived health gain. There are also some countries that have a higher QALY if a product is indicated for a rare or orphan disease.
Value and importance of real-world evidence for optimal reimbursement
While manufacturers have traditionally relied upon clinical trials for drug development, real-world evidence (RWE) has become a vital source of information in global reimbursement processes. RWE provides clinical evidence regarding a medical product’s safety and efficacy, derived from real-world data (RWD) collected during routine healthcare delivery.
RWE includes data derived from electronic health records, medical claims, product or disease registries, and other sources, such as digital health technologies. The key distinction is that this data is not collected in a controlled research environment; instead, it reflects how a drug performs in real-world conditions, providing a crucial foundation for evaluation.
The exponential increase in electronic data and improved analytics tools that are now available to all stakeholders will also boost reliance upon RWE for determining drug performance. New methodologies for leveraging RWE are expected to influence decision-making and enable greater understanding of how factors such as demographics, comorbidities, geography, and lifestyle can help healthcare providers decide the right product to prescribe for specific subpopulations, as is the case with precision medicine.
Furthermore, as manufacturers assess the value and comparative effectiveness of their products, they will also need to collect and use patient-reported outcomes (PROs) to enhance RWE generation. PROs are typically the best source of information about how a person is feeling and managing outside of the clinic, and can provide valuable information from the patient perspective on the effectiveness, safety, and tolerability of health interventions.
RWE documents value
Today, manufacturers, regulators, and payers increasing rely upon RWE to demonstrate the value of new drugs and gain a deeper understanding of their safety and effectiveness across diverse patient populations beyond the confines of clinical trials. Multiple survey articles indicate that RWE is now integrated throughout the product lifecycle, supporting both regulatory access and reimbursement submissions.
RWE plays an essential and increasingly prominent role in helping drug developers to demonstrate the value of their products to regulators. By providing data-driven insights into the drug's actual benefits and risks in everyday clinical settings, manufacturers can advance both clinical and financial objectives, highlighting competitive advantage for their products.
Real-time, RWD analysis enhances the understanding of specific diseases, supporting in the development of treatment approaches and identifying opportunities to support coverage decisions. Increasingly, HTA bodies are seeking RWE to help address uncertainty and generate more longitudinal data that verifies the durability of the clinical responses over time. This is particularly vital for securing optimal reimbursement, especially when clinical trial data alone may not be sufficient.
RWE is utilised from early opportunity assessments to launch planning, evidence generation, brand management, commercial optimisation, and post-marketing assessments. RWE supports regulatory decision-making in post-marketing surveillance to further document the product's safety profile and gather supplemental evidence needed to support ongoing market access and reimbursement.
Addressing pricing sustainability
Groundbreaking cell and gene therapies (CGTs) are offering new treatment options for patients with previously untreatable rare diseases and are set to transform the treatment landscape. As part of precision or personalised medicine, CGTs are designed to tailor treatments to a patient's specific genetic profile, environment, diet, and lifestyle. This approach enables a more targeted strategy for disease treatment based on each person’s unique characteristics.
However, these treatments are often associated with high upfront costs, largely associated with complex, time-consuming and costly manufacturing processes that carry requirements for specialised equipment, staff and facility costs, high-cost raw materials, and skilled labour. Collectively, these expenditures drive up the production costs and ultimately affect pricing and reimbursement strategies.
In response to the sometimes multi-million-dollar price tags associated with these novel, life-changing therapies, payers and biopharmaceutical manufacturers are increasingly engaging in value-based agreements and negotiating alternative payment models to improve affordability. These financial solutions connect reimbursement, coverage, or payment to the effectiveness and real-world performance of treatments over a specified time period.
Lower-cost biosimilars
With limited drug or health budgets, some industry analysts suggest that less expensive biosimilars may be the solution to free up finances for novel, more expensive agents. However, the potential for creating future biosimilar competition for CGTs to lower prices and improve patient access may be challenging. This is largely due to the complexity of CGTs, the regulatory requirement to demonstrate high similarity with no clinically meaningful differences, as well as challenges related to intellectual property and market size.
Other industry experts regard gene therapies as better candidates for biosimilar development than cell therapies. They assert that biosimilarity can be achieved when gene therapy biosimilars contain the same genetic sequence as a reference product, and the variability in the vector meets the high similarity standard.
Regulatory pathways and accelerated approvals
While there is no current international standard or regulatory framework for the approval of CGTs, there are expedited pathways such as priority review and accelerated assessment for CGT development. Many countries are introducing initiatives that are designed to support the efficient development of promising therapies:
- The US FDA, for example, prioritises CGTs for paediatric rare diseases that are difficult to study with randomised or placebo-controlled trials.
- EMA’s voluntary PRIME programme enhances support for the development of medicines that target an unmet medical need.
- Japan’s PMDA offers the Sakigake designation to accelerate innovative therapies addressing serious unmet medical needs, offering shorter lead times for regulatory consultation and faster new drug application reviews.
Ensuring patient accessibility
Innovation deserves recognition, for the substantial investments and risks that biopharmaceutical manufacturers take to develop groundbreaking treatments. By fostering innovation, agility, and collaboration, manufacturers can strengthen their commitment to improving the lives of patients with rare diseases and accelerating the development of life-improving therapies. The essential goal is to ensure that the benefits of these new treatments are accessible to patients.
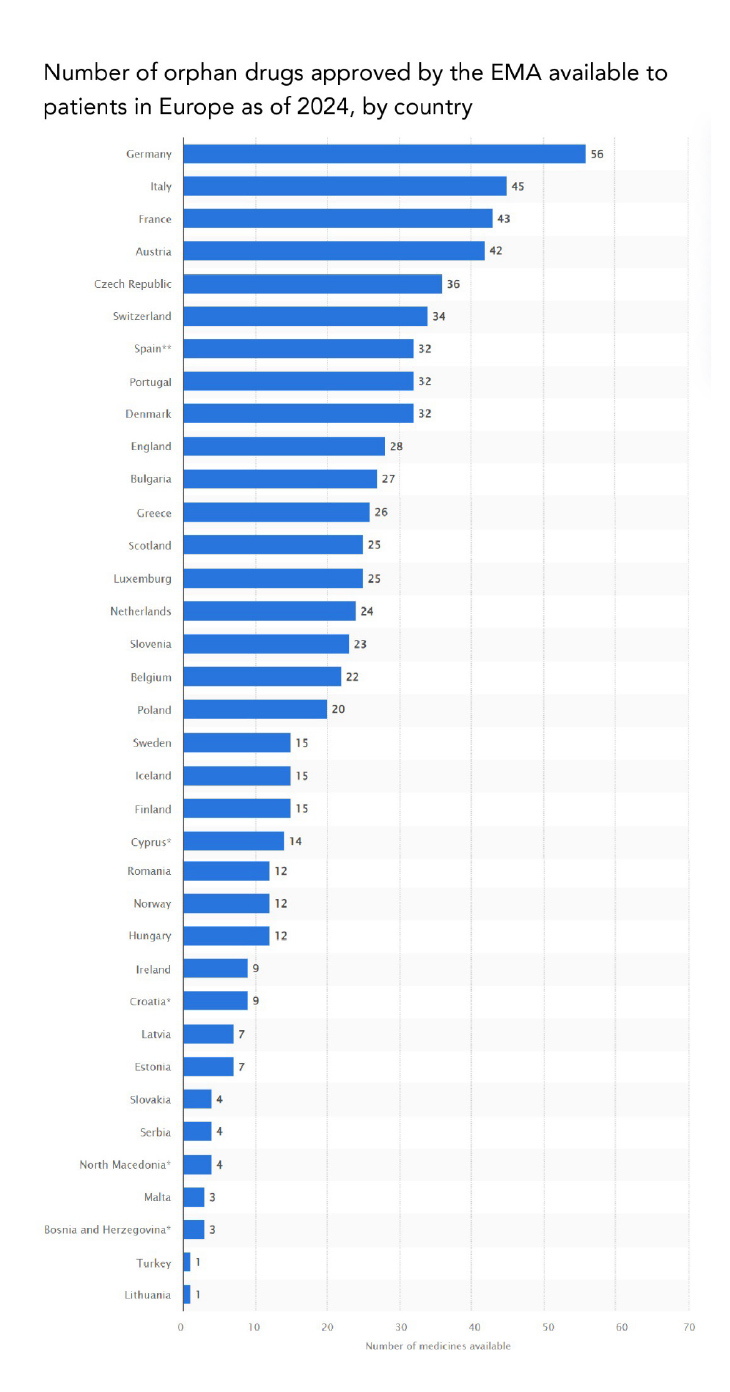
Source: STATISTA, 2024
https://www.statista.com/statistics/1248698/rate-of-orphan-drugs-availability-europe-by-country/
NOTE: Availability equates to reimbursement, with a few exceptions
The social impact of these medicines is palpable: patients stay in the workforce, contribute to society and have a better quality of life. Prior to the availability of these curative treatments, these lifestyle improvements would not be possible. Patients may have been unable to work because treatment was geared towards symptom management only, resulting in heavy utilisation of hospital emergency departments and in-patient care.
Looking ahead, global information sharing is crucial for accelerating drug development and deepening understanding of many rare diseases. Advancing the regulatory science needed to evaluate treatments for rare disease drugs will necessitate stronger collaboration between the FDA, EMA, and other leading agencies. Such global collaboration will expedite the development and approval of drugs targeting rare conditions, alleviating burdens on patients and caregivers while enhancing the quality of life for some of the world’s most vulnerable populations.
About the author

Gillian Molloy serves as VP of market access, EU/UK, at AscellaHealth. Molloy brings almost 20 years of experience in the life sciences industry to her role, in both the European and US markets. She has held commercial and market access leadership positions at Baxter, Novartis Oncology, and AstraZeneca, as well as trade relations and formulary strategy leadership roles at UnitedHealth Group. At AscellaHealth, Molloy provides strategic innovation and consultative market access support to pharmaceutical manufacturers and healthcare organisations. Prior to moving into the life sciences industry, Molloy held a chief pharmacist position in the Mater Misericordiae University Hospital, Dublin. She holds a BSc in Pharmacy and an MSc in Hospital Pharmacy from the University of Dublin, Trinity College, as well as an MBA from University College Dublin Michael Smurfit Graduate Business School. Molloy also has a Diploma in Health Economics from the National University of Ireland Whitaker School of Government and Management.









