Rare diseases and orphan drugs need digital innovation

More patient-centric study design, using mHealth for longer-term monitoring, will address many of the inherent challenges in conducting pivotal clinical trials in rare diseases, argues Elin Haf Davies.
Pharma companies are leveraging digital health data more than ever before to improve clinical trials, in a bid to develop drugs that could have a significant impact on patients’ lives.
No area could benefit more from this disruptive approach than the rare disease sector and the development of orphan drugs: an area of high unmet therapeutic need and large financial investment.
There are estimated to be between 6,000 and 8,000 rare diseases affecting up to 60 million people across Europe and the US. The orphan drug sector is expected to experience strong growth over the coming years; EvaluatePharma (2014) estimates total orphan drug sales of $176 billion by 2020. There were 2,476 ongoing national or international clinical trials for 629 rare diseases in 29 countries (IDiRC/Orphanet) in 2014.
EvaluatePharma (2015) estimates that the average phase 3 clinical trial for an orphan drug costs $103 million and suggests that, based on the current pipeline of phase 3 drugs in development for orphan drugs, the total spent by pharma companies will be $6.9 billion on phase 3 trials alone.
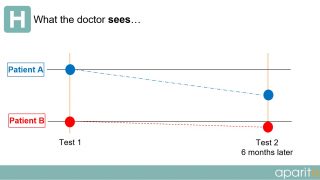
In current clinical trials, a patient visits a doctor or researcher at fixed intervals during the trial and data is taken from them. However, as depicted in the diagrams below, a large amount of important information is completely missed or not observed during routine testing.
What the doctor sees:
What the patient experiences:
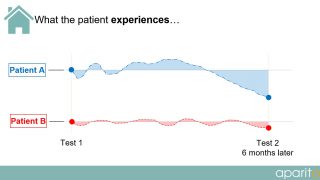
The choices of endpoints and outcome measures in many trials do not reflect the issues that are important to patients living their day-to-day lives. What patients consider relevant is an important criterion and, as such, patient perspectives on other aspects of their disease/treatment experience that are significant to other stakeholders should not be ignored.
In rare disease, where patient numbers are low, and both disease presentation and rate of progression are heterogeneous, relying on data from sterile snapshot assessments during clinic visits alone leads to inconclusive trial results, which is both unethical and uneconomical.
As a result of poor information gathered during clinical trials, a significant number of them fail – especially those involving children – at significant cost to the pharma industry. Recent research suggests that 42% of recently completed paediatric trials failed to establish safety and efficacy (Wharton, G T et al [2014]) and, out of 103 orphan drugs approved by the EU, 37% failed at the Health Technology Assessment (HTA) stage owing to insufficient effectiveness information for the payer (Brook, A. PPi Healthcare Consulting [2015]).
In light of these challenges, regulators and payers are increasingly demanding real-world data that demonstrates both the efficacy and the effectiveness of the drug. This is echoed in the recent adaptive pathway pilots conducted by the European Medicines Agency.
An obvious solution is to use the digital health revolution and tap into the global availability of smartphones and the rapid improvements in the size, appearance, usability, battery life and costs of wearables.
The regulatory and reimbursement pathways for mHealth and wearables are not clear, however – not only in terms of understanding which regulatory routes and demands apply for medical devices and apps, but also in understanding how generated data will be analysed by regulators and payers for drug evaluation.
Throughout Europe, standalone software and apps that meet the definition of a medical device must still be CE marked, in line with the EU medical device directives, to ensure they are regulated and acceptably safe to use and also perform as the manufacturer/developer intends.
Consumer–grade trackers
However, as of June 2016, there were 104 completed, current or pending Fitbit studies on the ClinicalTrials.gov website, including studies in obesity, diabetes and cancer. Consumer-grade activity trackers, such as Fitbit, are not considered medical devices, and it is not yet clear how regulators will respond to the data generated. However their low cost and consumer-friendly functions make them a popular choice for pharma and patients alike.
Given the fact that patients’ wellbeing between clinic visits is based on no objective data at all, and on what the patient can remember, does the fact that a Fitbit may record 70 steps when a patient has actually only taken 68 really matter? Will consistently misreading a heart rate by 2-3 beats per minute be important in chronic diseases?
Recent challenges encountered with using the Six-Minute Walk Test as a primary endpoint in Duchenne studies further highlight this issue. Is 12-18 minutes’-worth of data from a two-year study, generated during stressful and sterile hospital visits, really considered superior to continuous data captured in real time throughout the patient’s trial experience using a consumer-grade wearable?
I would argue not.
The current drug development pathway for rare diseases is not sustainable, either in terms of addressing the high unmet therapeutic needs of patients through ensuring rapid patient access to innovative drugs, or in providing orphan drugs that are affordable to current healthcare economies.
A more patient-centric study design will address many of the inherent challenges in conducting pivotal clinical trials in rare diseases, allowing:
- fewer hospital visits,
- less emphasis on patient memory recall during clinics
- more depth in the data collected for each individual patient
- no paper documentation and manual entries
- access to real-time data which provides the ability to make earlier decisions.
Objective data, captured passively and in real time with wearable technology, and contextualised with patient data input linked to medication adherence, side effects/adverse events and Patient Reported Outcomes, provides the platform to capture and analyse in-depth and meaningful patient data. Providing such a platform in accordance with regulatory and governance demands is one step towards enabling an adaptive and iterative pathway to drug development, such as that promoted by the enabling platform ADAPT SMART (see www.adaptsmart.eu)
ADAPT SMART works to accelerate development of appropriate patient therapies in a sustainable way. By assisting the coordination of Medicines Adaptive Pathways to Patients (MAPPs), it seeks to foster access to beneficial treatments for the right patient group at the earliest appropriate time in the product’s lifecycle. Providing drugs at this stage introduces some uncertainty, which, in turn, will require a higher level of patient monitoring – but one that is conducive with patients’ daily lives.
Many rare diseases are likely to meet the engagement criteria set for MAPPs. Introducing digital health innovations in this field will facilitate both the need for increased patient monitoring and the generation of real-world evidence.
Our studies in India, America and the UK, for example, introducing wearables and disease-specific apps for proof-of-concept and observational studies in Gaucher disease, Niemann-Pick C and Late Onset Tay Sachs, are based on such needs, and have been instigated in partnership with patient groups, physicians and pharma.
About the author:
Elin Haf Davies PhD began her clinical career at Great Ormond Street Hospital in London, UK. After 11 years of clinical, research and academic experience, she went on to work in the Paediatric Team at the European Medicines Agency, implementing the Paediatric Regulation in Europe. Six years later she left to become an Associate Fellow at CASMI (Oxford University) and to found aparito.
Read more on rare diseases:
Tunnah’s musings: Rare diseases hold key to future of drug development